suppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="/c/Users/Saideep/Box/imlab-data/data-Github/Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="replicating-kircher-mutagenesis"## copy the slug from the headerbDATE='2023-03-13'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))
[1] 1
Code
WORK=DATA
Context
Last week we implemented reverse complement prediction into our core, merged Enformer pipeline after correcting a different bug we had with it. The reverse complement still appears to not be working as intended, so we are going to debug it today. Once this is done, I can rerun the Kircher mutagenesis for LDLR and see if it replicates findings from another review paper.
In the meantime, I have also been rerunning the full GEUVADIS set of individuals across all protein coding genes. With those complete, I am rerunning the quantification steps and generating new expression tables. With these, I can finally rerun the portability, ERAP2, and epistasis analyses to confirm the prior findings.
Quantifying GEUVADIS expression data
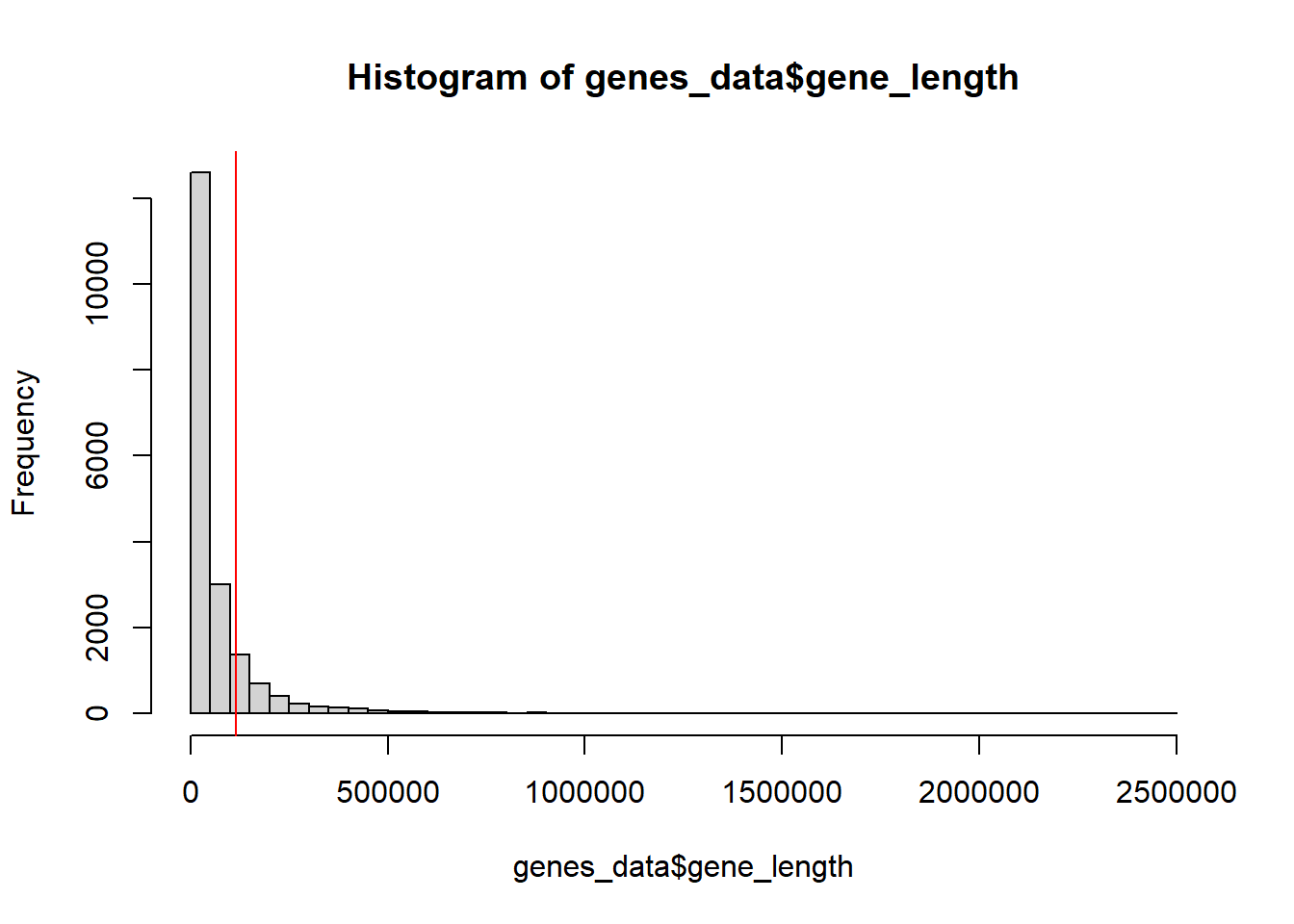
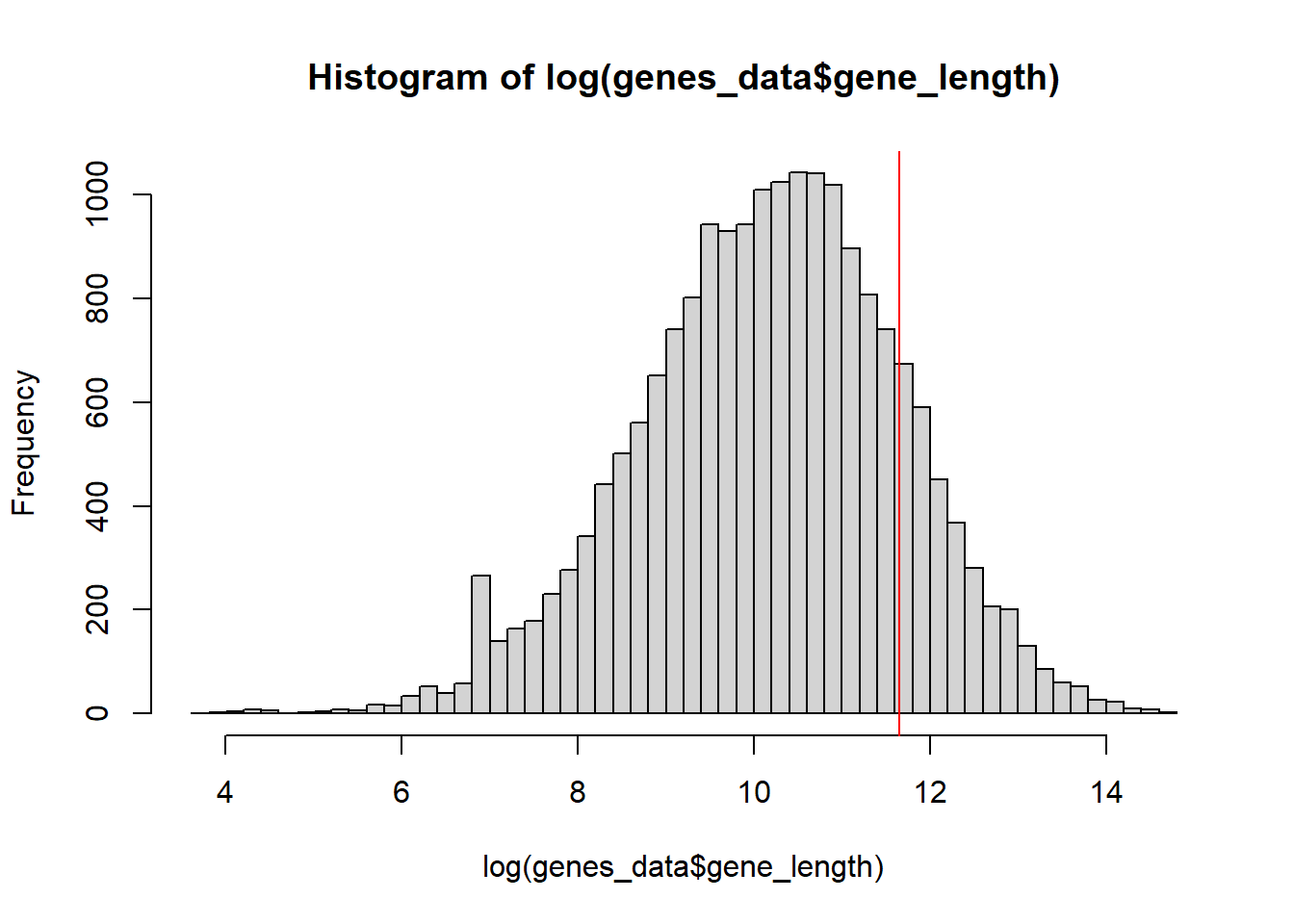
One thing I noticed is that some genes are larger than Enformer’s output window size (114688). In these cases, quantification doesn’t really work. These genes tend to error out.
As can be seen, a significant number of genes(16%) are too large for Enformer’s window size. These will be ignored in the final quantification. It should also be noted that many genes are also lacking in cis-regulatory context because they are close in size to the window size. It may be worth taking note of these in the future.
Speeding up quantification
My quantification is currently parallelized by target region (in this case genes). The easiest way to speed up the quantification is to use more cores at once. On thetagpu, each node has 2 64 core cpus. We can therefore utilize 128 cores by running on the node.
Code
qsub-I-n 1 -t 1:00:00 -q single-gpu -A covid-ct
This runs reasonably fast (~1,2 hours). For larger jobs, however, we probably want to parallelize on thetacpu.
This blog post is somewhat empty because the GEUVADIS data goes into dedicated analyses rather than daily blog posts.
Replicating Kircher Mutagenesis
Due to time constraints and focusing on re-quantifying the Geuvadis results, the Kircher replication is delayed a bit. The reverse complement issue is first on tomorrow’s priority list.
Source Code
---title: "Replicating Kircher Mutagenesis"author: "Saideep Gona"date: "2023-03-13"format: html: code-fold: true code-summary: "Show the code"execute: freeze: true warning: false---```{r}#| label: Set up box storage directorysuppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="/c/Users/Saideep/Box/imlab-data/data-Github/Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="replicating-kircher-mutagenesis"## copy the slug from the headerbDATE='2023-03-13'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA```# ContextLast week we implemented reverse complement prediction into our core, merged Enformer pipeline after correcting a different bug we had with it. The reverse complement still appears to not be working as intended, so we are going to debug it today. Once this is done, I can rerun the Kircher mutagenesis for LDLR and see if it replicates findings from another review paper.In the meantime, I have also been rerunning the full GEUVADIS set of individuals across all protein coding genes. With those complete, I am rerunning the quantification steps and generating new expression tables. With these, I can finally rerun the portability, ERAP2, and epistasis analyses to confirm the prior findings.## Quantifying GEUVADIS expression dataOne thing I noticed is that some genes are larger than Enformer's output window size (114688). In these cases, quantification doesn't really work. These genes tend to error out.The following code explores this:```{r}library(tidyverse)gene_info_file <-file.path(DATA,"protein_coding_nosex.bed")genes_data <-read_delim("protein_coding_nosex.bed", col_names =FALSE)colnames(genes_data) <-c("chr","start","end","gene_id","gene_name")genes_data$gene_length <- genes_data$end - genes_data$start``````{r}enformer_window =114688hist(genes_data$gene_length, breaks =50)abline(v=114688, col ="red")hist(log(genes_data$gene_length), breaks =50)abline(v=log(114688), col ="red")sum(genes_data$gene_length > enformer_window)sum(genes_data$gene_length > enformer_window)/nrow(genes_data)```As can be seen, a significant number of genes(16%) are too large for Enformer's window size. These will be ignored in the final quantification. It should also be noted that many genes are also lacking in cis-regulatory context because they are close in size to the window size. It may be worth taking note of these in the future.## Speeding up quantificationMy quantification is currently parallelized by target region (in this case genes). The easiest way to speed up the quantification is to use more cores at once. On thetagpu, each node has 2 64 core cpus. We can therefore utilize 128 cores by running on the node. ```{bash, eval=FALSE}qsub-I-n 1 -t 1:00:00 -q single-gpu -A covid-ct```This runs reasonably fast (~1,2 hours). For larger jobs, however, we probably want to parallelize on thetacpu.This blog post is somewhat empty because the GEUVADIS data goes into dedicated analyses rather than daily blog posts.## Replicating Kircher MutagenesisDue to time constraints and focusing on re-quantifying the Geuvadis results, the Kircher replication is delayed a bit. The reverse complement issue is first on tomorrow's priority list.