suppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="C:\\Users\\Saideep\\Box\\imlab-data\\data-Github\\Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="replicating-kircher-mutagenesis-p2"## copy the slug from the headerbDATE='2023-03-14'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA
Context
Last week we implemented reverse complement prediction into our core, merged Enformer pipeline after correcting a different bug we had with it. The reverse complement still appears to not be working as intended, so we are going to debug it today (delayed from yesterday). Once this is done, I can rerun the Kircher mutagenesis for LDLR and see if it replicates findings from another review paper.
In addition, yesterday I quantified the GEUVADIS results and did some experimenting with faster ways of doing the quantification. Today I will also start updating the ERAP2, Portability, and Epistasis analyses.
Kircher Mutagenesis
Fixing reverse compliment prediction in merged pipeline
Temi and I figured out the source of the bug with the merged pipeline. The following code reverse-compliments a 2-D one-hot sequence:
Code
np.flip(np.flip(sequence_one_hot,axis=1),axis=0)
However, in the merged pipeline Temi had been working with a 3-D one-hot sequence where the 0th axis could be used for batching. After revision, the following code resolved the issue:
Code
np.flip(np.flip(sequence_one_hot,axis=1),axis=2)
Replicating variant effect predictions
After resolving issues with the merged pipeline, I reran the Kircher mutagenesis analysis. The process is to compute the promoter region expression in both the forward and reverse commpliments for each variant. Then the average between the forward and reverse is used as basal expression and the fold-change between reference and alternative is calculated. This is almost an exact replica of the methodology used here: https://www.biorxiv.org/content/10.1101/2022.09.15.508087v1.full.pdf. The only major differences are:
Indels are not considered
Input sequences are not centered at the variants being mutated, but rather are fixed to the gene of interest (in this case LDLR)
I don’t have the exact data used to generate the reference variant effect sizes from the review paper, just data from the original Kircher mutagenesis study linked below. Unfortunately, the log2 variant effects dont seem to exactly match the values used in the above review (the range of variant effect sizes seems different). It’s somewhat tough to trace why this is. The link to the data is here: https://osf.io/75b2m/
Nevertheless, my results do correlate with the Kircher variant effects and look roughly similar to the review study figure. Results shown below:
Enformer vs. Kircher from Review Paper
Enformer vs. Kircher from Our Pipeline
As you can see, the results are quite close but not identical. Considering the review paper authors asked the Kircher authors directly for variant effect data, I’m not sure if its worth digging deeper. We have also done pretty extensive manual checks of our pipeline correctness at this point, so bugs are unlikely at this point.
Adding main analyses to lab folders
Similar to these daily blog posts, it is also worth adding my main analyses to the common lab github + Box system. To this end, I’ve added the following directory to the lab Box: “imlab-data/data-Github/analysis-Sai/” which will store data tables, etc. from analysis. There is a also a new github repository at “https://github.com/hakyimlab/analysis-Sai” which is also a quarto website similar to these blog posts. I will be posting updates to long running analyses here.
Source Code
---title: "Replicating Kircher Mutagenesis p2"author: "Saideep Gona"date: "2023-03-14"format: html: code-fold: true code-summary: "Show the code"execute: freeze: true warning: false---```{r}#| label: Set up box storage directorysuppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="C:\\Users\\Saideep\\Box\\imlab-data\\data-Github\\Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="replicating-kircher-mutagenesis-p2"## copy the slug from the headerbDATE='2023-03-14'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA```# ContextLast week we implemented reverse complement prediction into our core, merged Enformer pipeline after correcting a different bug we had with it. The reverse complement still appears to not be working as intended, so we are going to debug it today (delayed from yesterday). Once this is done, I can rerun the Kircher mutagenesis for LDLR and see if it replicates findings from another review paper.In addition, yesterday I quantified the GEUVADIS results and did some experimenting with faster ways of doing the quantification. Today I will also start updating the ERAP2, Portability, and Epistasis analyses.## Kircher Mutagenesis### Fixing reverse compliment prediction in merged pipelineTemi and I figured out the source of the bug with the merged pipeline. The following code reverse-compliments a 2-D one-hot sequence:```{python, eval=FALSE}np.flip(np.flip(sequence_one_hot,axis=1),axis=0)```However, in the merged pipeline Temi had been working with a 3-D one-hot sequence where the 0th axis could be used for batching. After revision, the following code resolved the issue:```{python, eval=FALSE}np.flip(np.flip(sequence_one_hot,axis=1),axis=2)```### Replicating variant effect predictionsAfter resolving issues with the merged pipeline, I reran the Kircher mutagenesis analysis. The process is to compute the promoter region expression in both the forward and reverse commpliments for each variant. Then the average between the forward and reverse is used as basal expression and the fold-change between reference and alternative is calculated. This is almost an exact replica of the methodology used here: https://www.biorxiv.org/content/10.1101/2022.09.15.508087v1.full.pdf. The only major differences are:* Indels are not considered* Input sequences are not centered at the variants being mutated, but rather are fixed to the gene of interest (in this case LDLR)* I don't have the exact data used to generate the reference variant effect sizes from the review paper, just data from the original Kircher mutagenesis study linked below. Unfortunately, the log2 variant effects dont seem to exactly match the values used in the above review (the range of variant effect sizes seems different). It's somewhat tough to trace why this is. The link to the data is here: https://osf.io/75b2m/Nevertheless, my results do correlate with the Kircher variant effects and look roughly similar to the review study figure. Results shown below: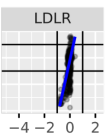{width=100}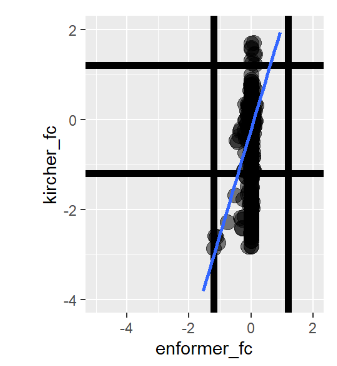{width=300}As you can see, the results are quite close but not identical. Considering the review paper authors asked the Kircher authors directly for variant effect data, I'm not sure if its worth digging deeper. We have also done pretty extensive manual checks of our pipeline correctness at this point, so bugs are unlikely at this point.## Adding main analyses to lab foldersSimilar to these daily blog posts, it is also worth adding my main analyses to the common lab github + Box system. To this end, I've added the following directory to the lab Box: "imlab-data/data-Github/analysis-Sai/" which will store data tables, etc. from analysis. There is a also a new github repository at "https://github.com/hakyimlab/analysis-Sai" which is also a quarto website similar to these blog posts. I will be posting updates to long running analyses here.

