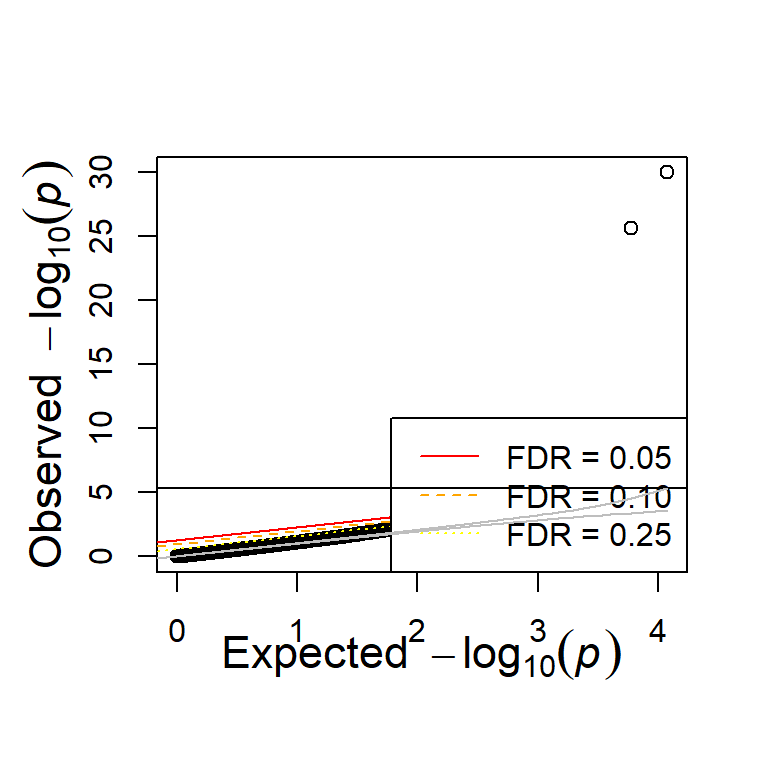suppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="C:\\Users\\Saideep\\Box\\imlab-data\\data-Github\\Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="updating-existing-geuvadis-analysis-p3"## copy the slug from the headerbDATE='2023-03-20'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA
Context
I still need to update my epistasis analysis with the new data. After doing so, the results are roughly as follows (full analysis is in the analysis repo).
Epistasis Results
Enrichment of epistatic signal
Like before, there is enrichment in both the additive and interaction test.
GSEA results
thesholds
This first plot shows the number of genes in background (significant for the additive test) and the number of genes in the enrichment set (significant for both additive and interaction tests) at varying thresholds.
gsea
The second plot shows GSEA results using the hallmark genesets and fgsea for the above gene sets. Like before, the results are null. This being said, I know Karl Tayeb in Mathew Stephens lab has been working on a different method for gene set enrichment which might be interesting. I am in contact with home to try and use his method.
In addition, the ribosomal genes I observed previously are no longer significant, so they may have been a relic of bugs I have since fixed.
Sasse et al. Preprint (https://www.biorxiv.org/content/10.1101/2023.03.16.532969v1.full.pdf)
It seems like others were also trying to understand why Enformer predicts negative predictions. Interestingly, they ended up going in a similar direction of looking at the mutagenesis profile for certain genes vs. the known causal variants. In this case, they also looked at the gradient effect (which input sites have the strongest impact on the outcome), and found that often times for the negative correlations Enformer is just misattributing the effect to different variants than the causal. It especially seems attracted to variants closer to promoter sites, which is consistent with this preprint: https://www.biorxiv.org/content/10.1101/2022.09.15.508087v1.full.
This approach is very similar to the direction I was planning to test in from the post “2023-03-10-implementing-personalized-mutagenesis”. There, I planned to do “personalized mutagenesis” to determine which variants are contributing to the personalized variation. I think we should also store the “contribution scores” from our runs for this purpose. This exploration can still compliment the Sasse et al. results by:
Confirming/extending in a different population + cell type + gene
Clarifying and quantifying the difference between In-silico Mutagenesis (ISM) and Personalized In-silico Mutagenesis (PISM)
Hackathon Prep
I watched the Andrej Karpathy tutorial video today, can start helping Temi prepare for the Hackathon tomorrow.
Source Code
---title: "Updating Existing GEUVADIS Analyses part 3"author: "Saideep Gona"date: "2023-03-20"format: html: code-fold: true code-summary: "Show the code"execute: freeze: true warning: false---```{r}#| label: Set up box storage directorysuppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="C:\\Users\\Saideep\\Box\\imlab-data\\data-Github\\Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="updating-existing-geuvadis-analysis-p3"## copy the slug from the headerbDATE='2023-03-20'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA```# ContextI still need to update my epistasis analysis with the new data. After doing so, the results are roughly as follows (full analysis is in the analysis repo).## Epistasis Results### Enrichment of epistatic signal 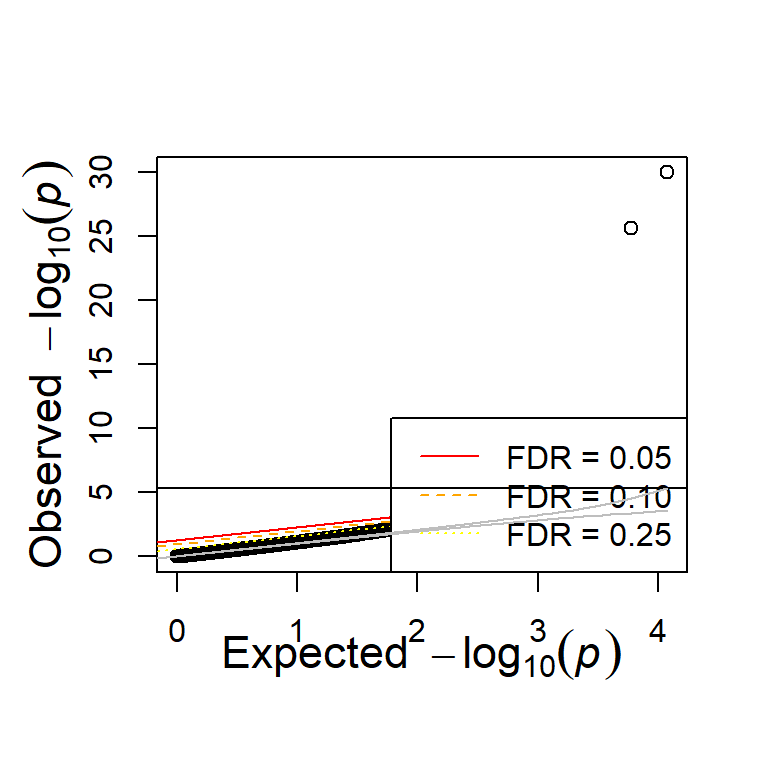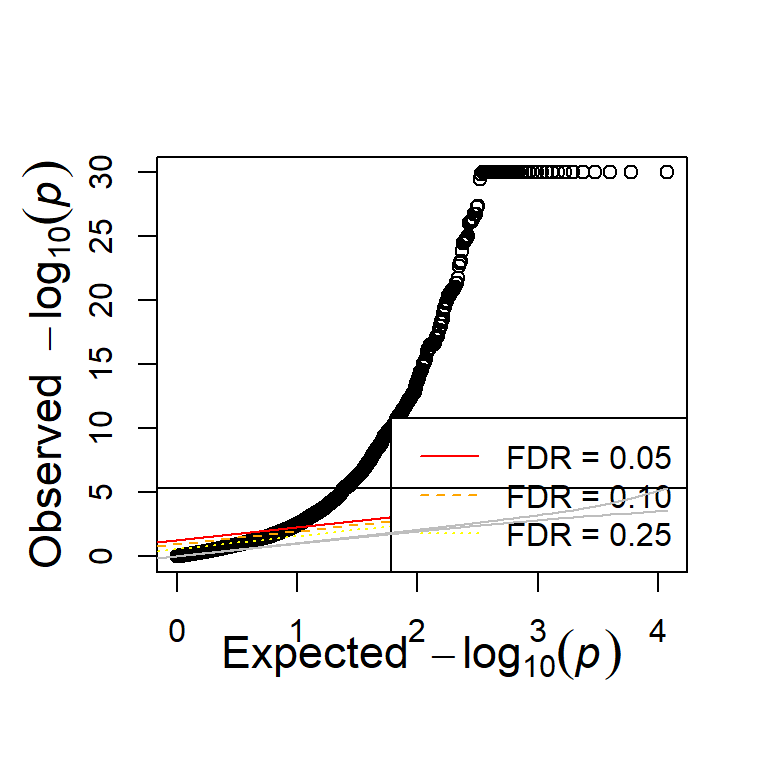Like before, there is enrichment in both the additive and interaction test. ### GSEA results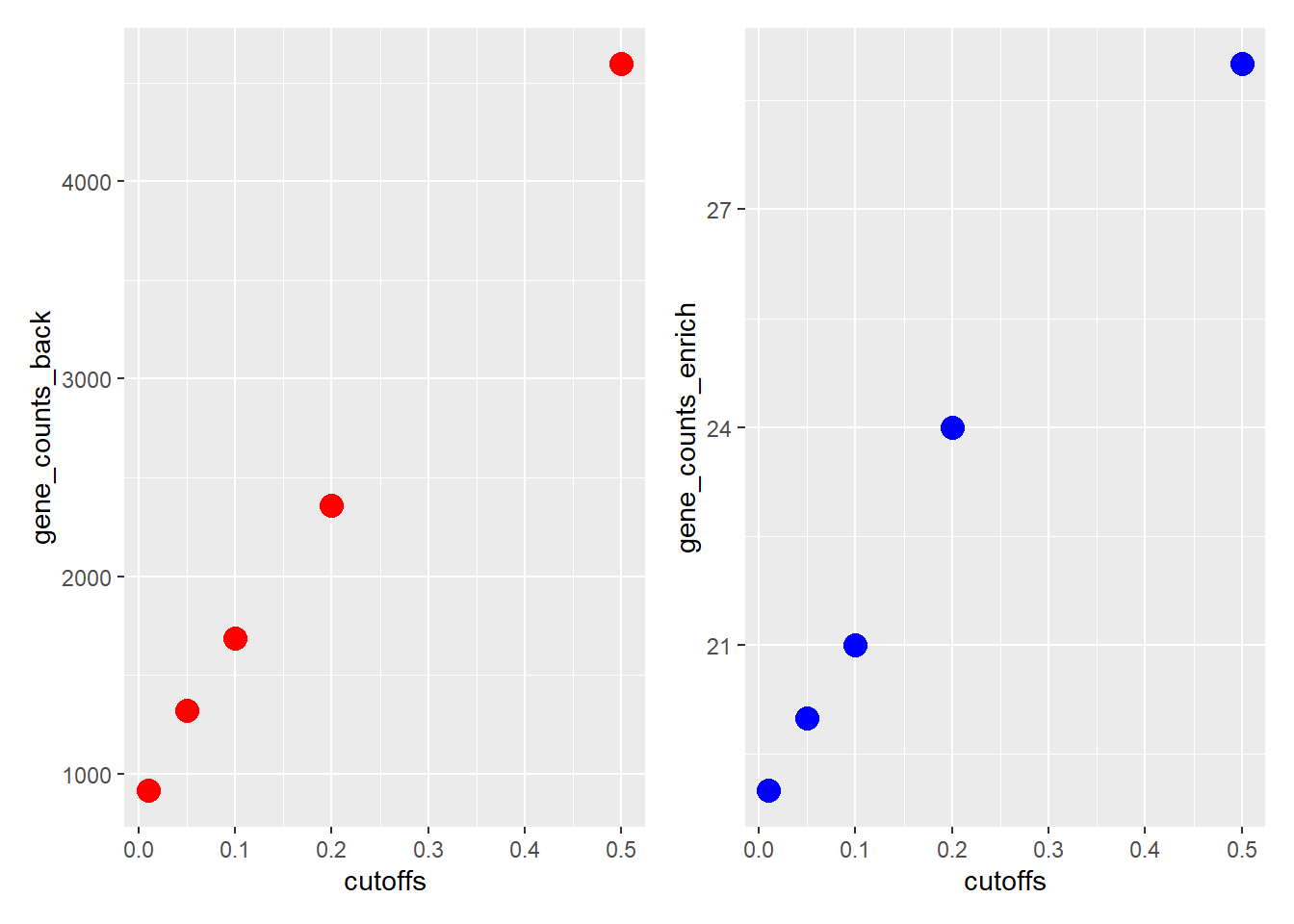This first plot shows the number of genes in background (significant for the additive test) and the number of genes in the enrichment set (significant for both additive and interaction tests) at varying thresholds.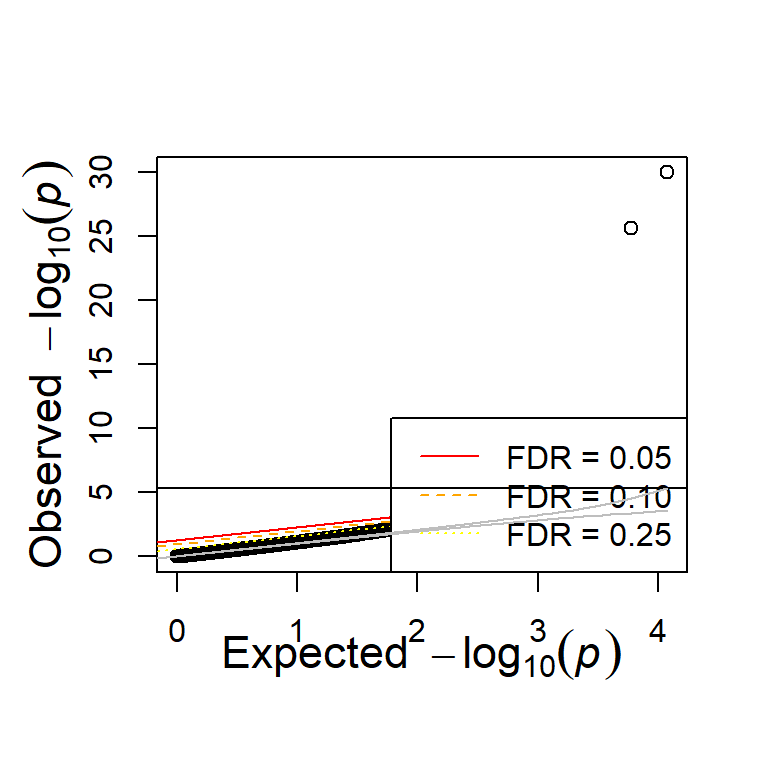The second plot shows GSEA results using the hallmark genesets and fgsea for the above gene sets. Like before, the results are null. This being said, I know Karl Tayeb in Mathew Stephens lab has been working on a different method for gene set enrichment which might be interesting. I am in contact with home to try and use his method.In addition, the ribosomal genes I observed previously are no longer significant, so they may have been a relic of bugs I have since fixed.## Sasse et al. Preprint (https://www.biorxiv.org/content/10.1101/2023.03.16.532969v1.full.pdf)It seems like others were also trying to understand why Enformer predicts negative predictions. Interestingly, they ended up going in a similar direction of looking at the mutagenesis profile for certain genes vs. the known causal variants. In this case, they also looked at the gradient effect (which input sites have the strongest impact on the outcome), and found that often times for the negative correlations Enformer is just misattributing the effect to different variants than the causal. It especially seems attracted to variants closer to promoter sites, which is consistent with this preprint: https://www.biorxiv.org/content/10.1101/2022.09.15.508087v1.full. This approach is very similar to the direction I was planning to test in from the post "2023-03-10-implementing-personalized-mutagenesis". There, I planned to do "personalized mutagenesis" to determine which variants are contributing to the personalized variation. I think we should also store the "contribution scores" from our runs for this purpose. This exploration can still compliment the Sasse et al. results by:* Confirming/extending in a different population + cell type + gene* Clarifying and quantifying the difference between In-silico Mutagenesis (ISM) and Personalized In-silico Mutagenesis (PISM)## Hackathon PrepI watched the Andrej Karpathy tutorial video today, can start helping Temi prepare for the Hackathon tomorrow.