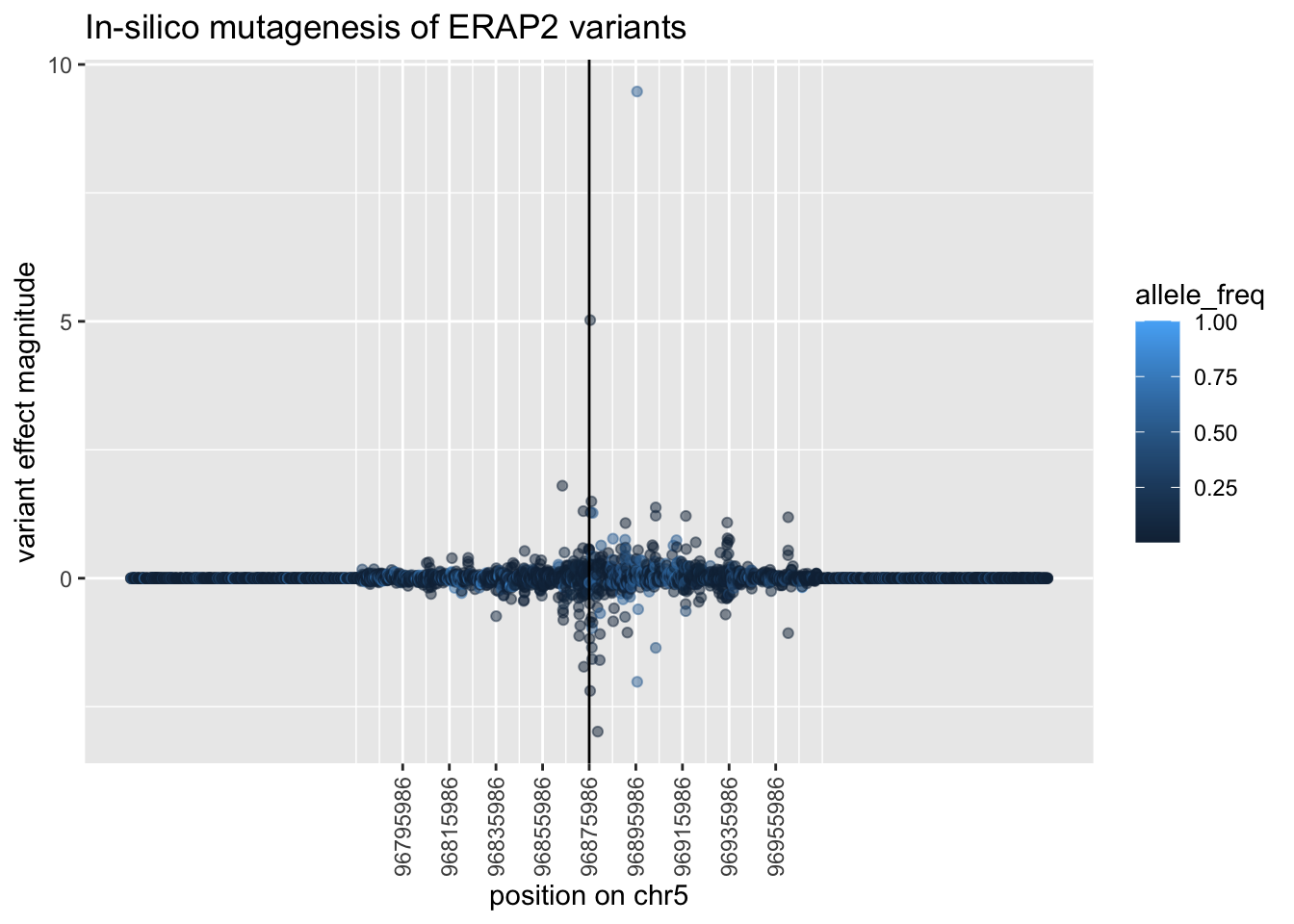
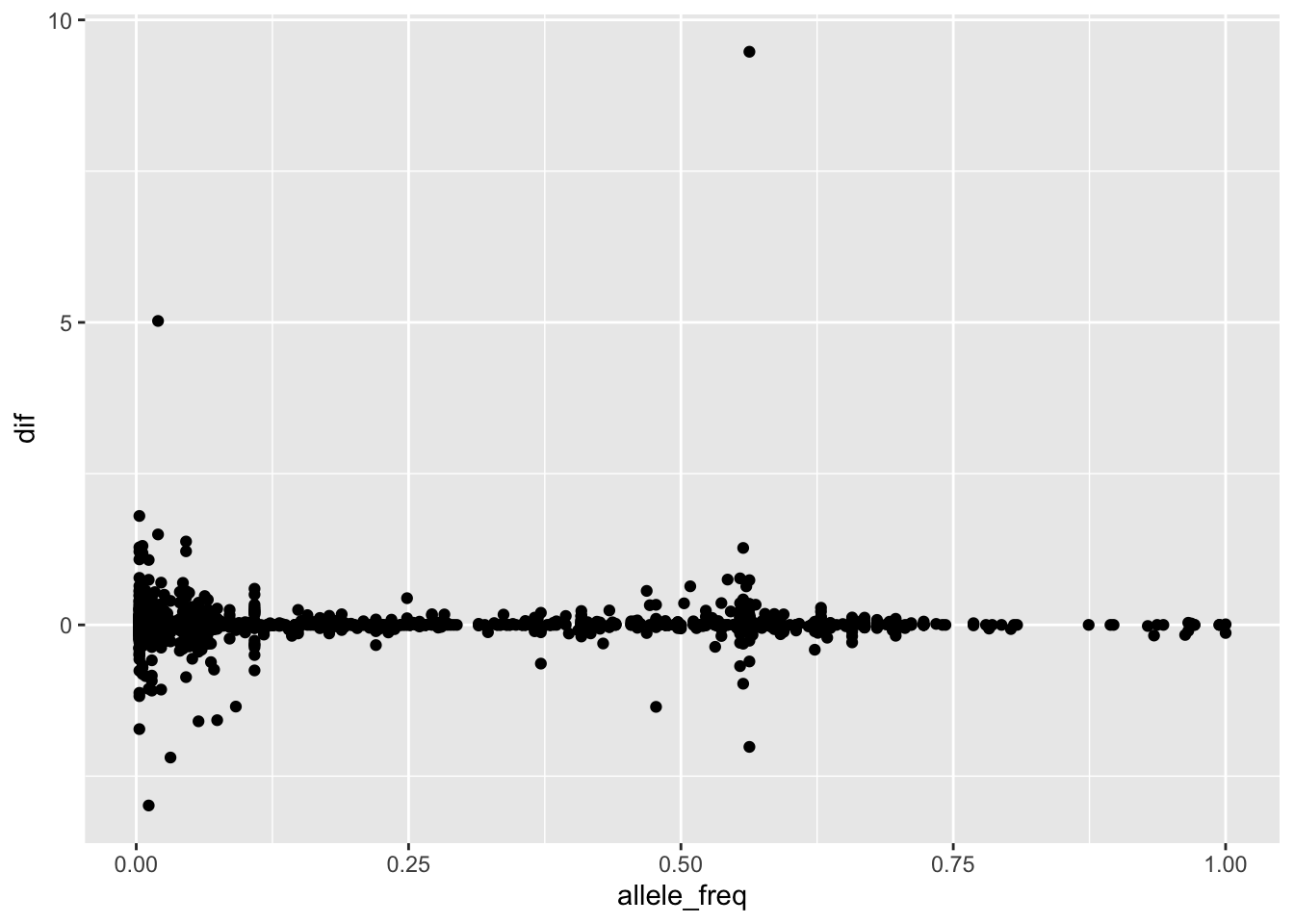
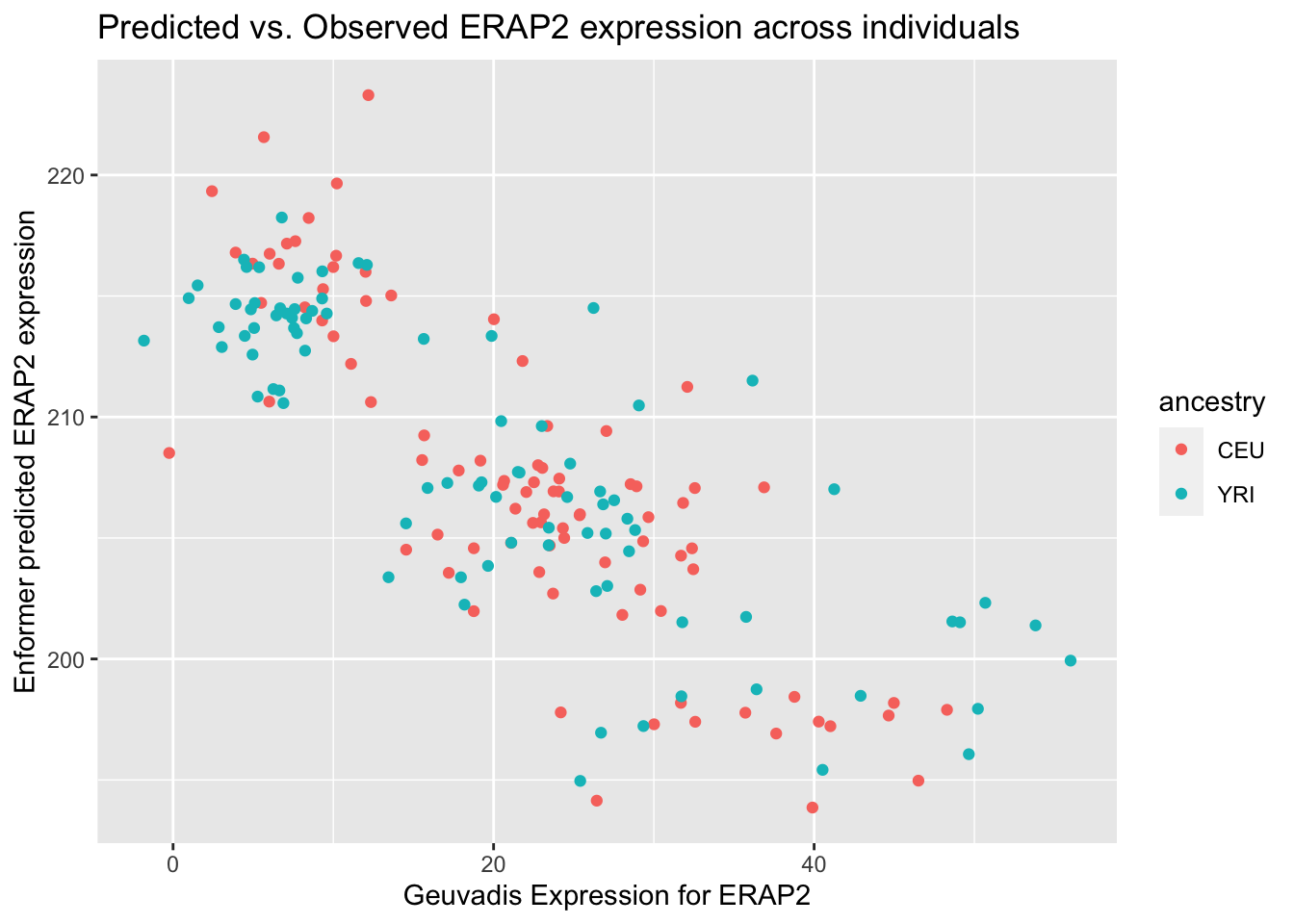
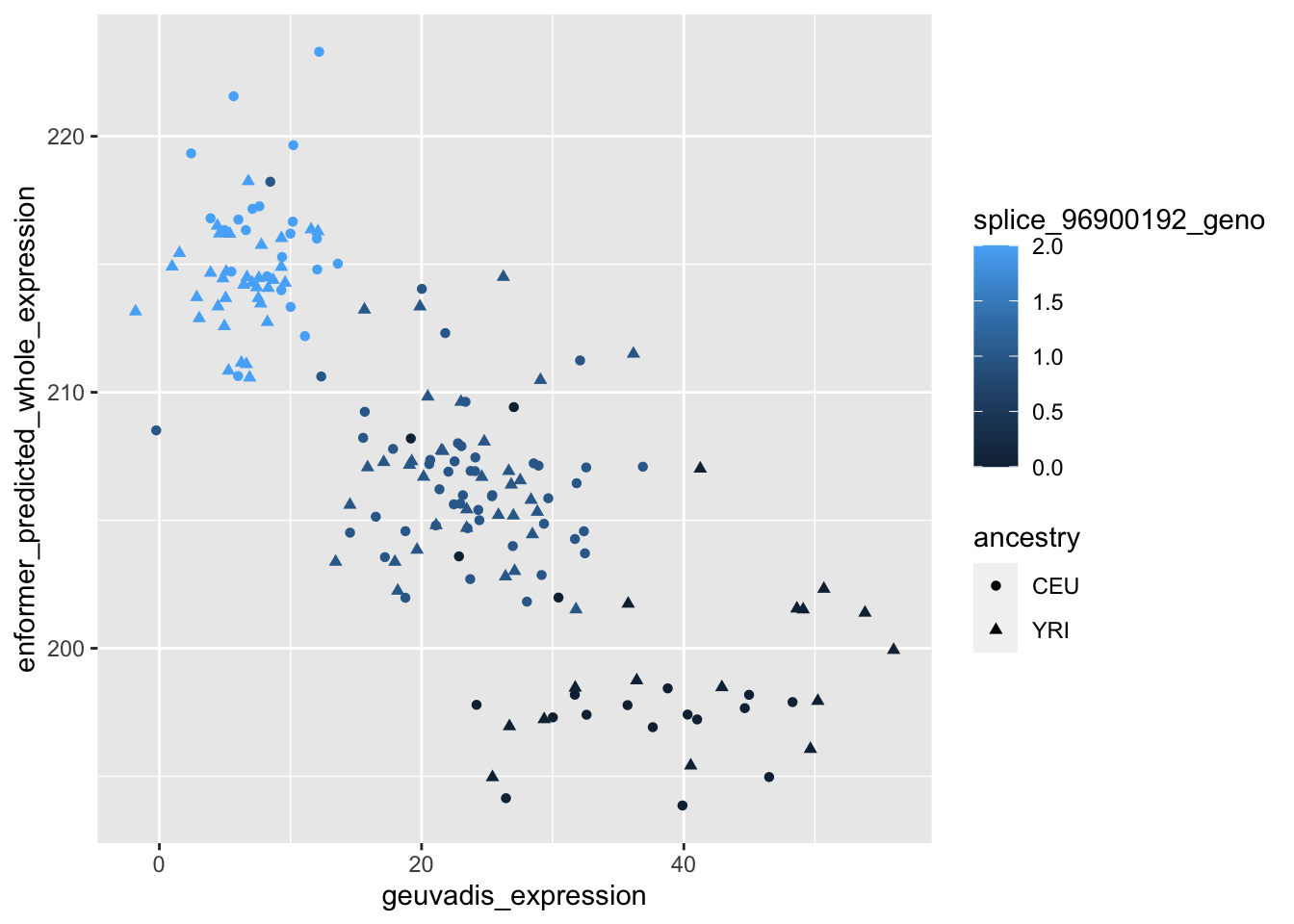
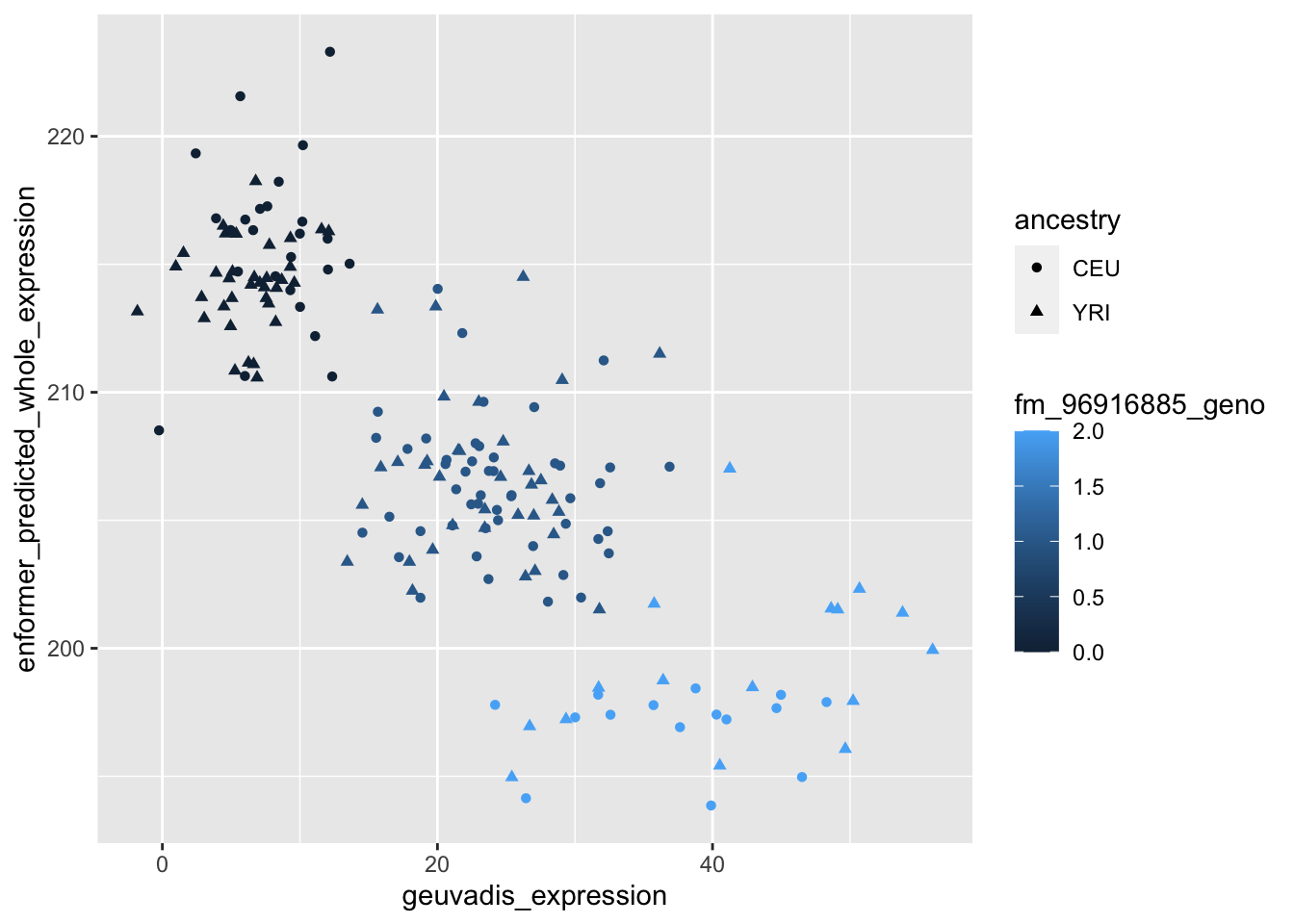
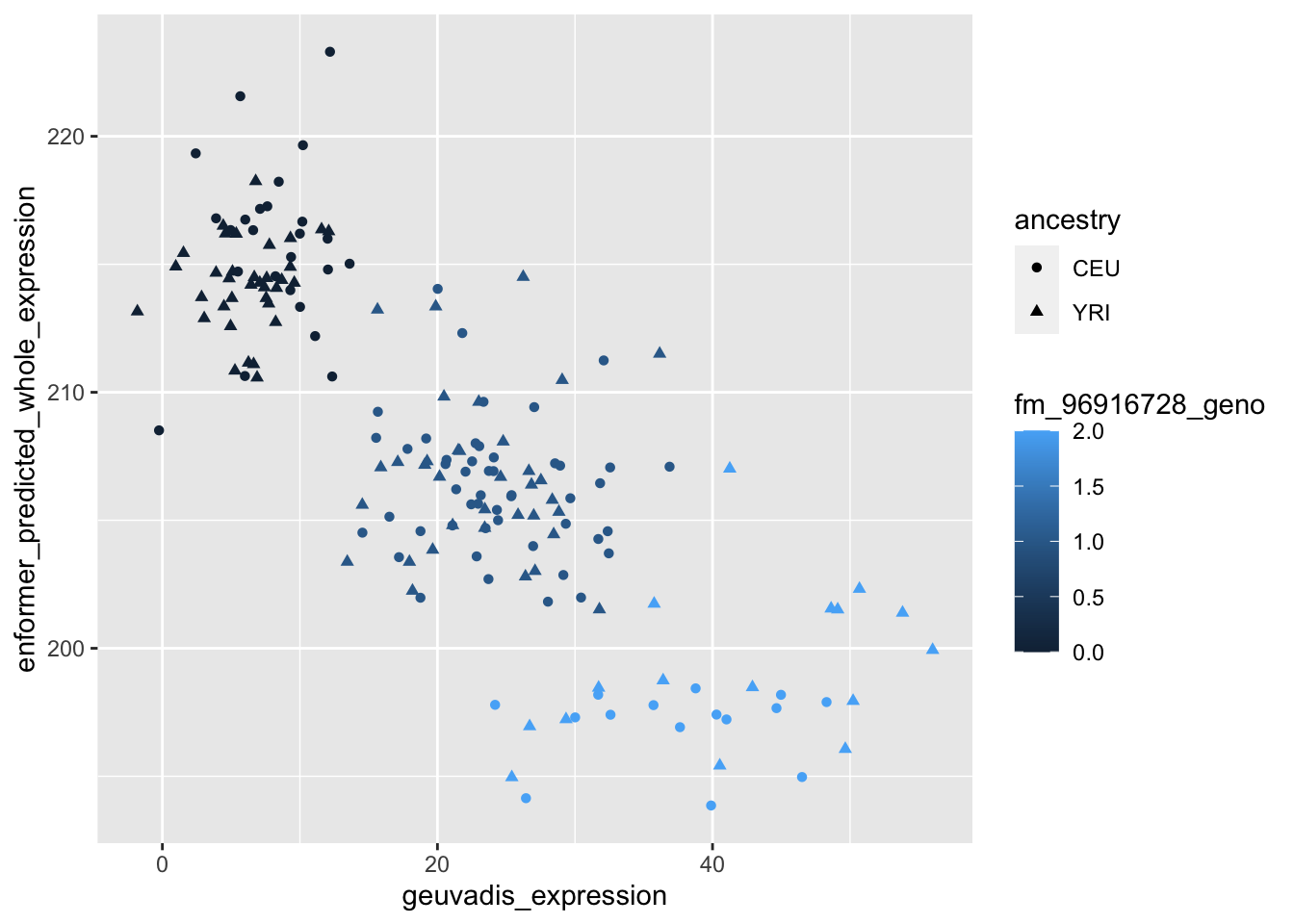
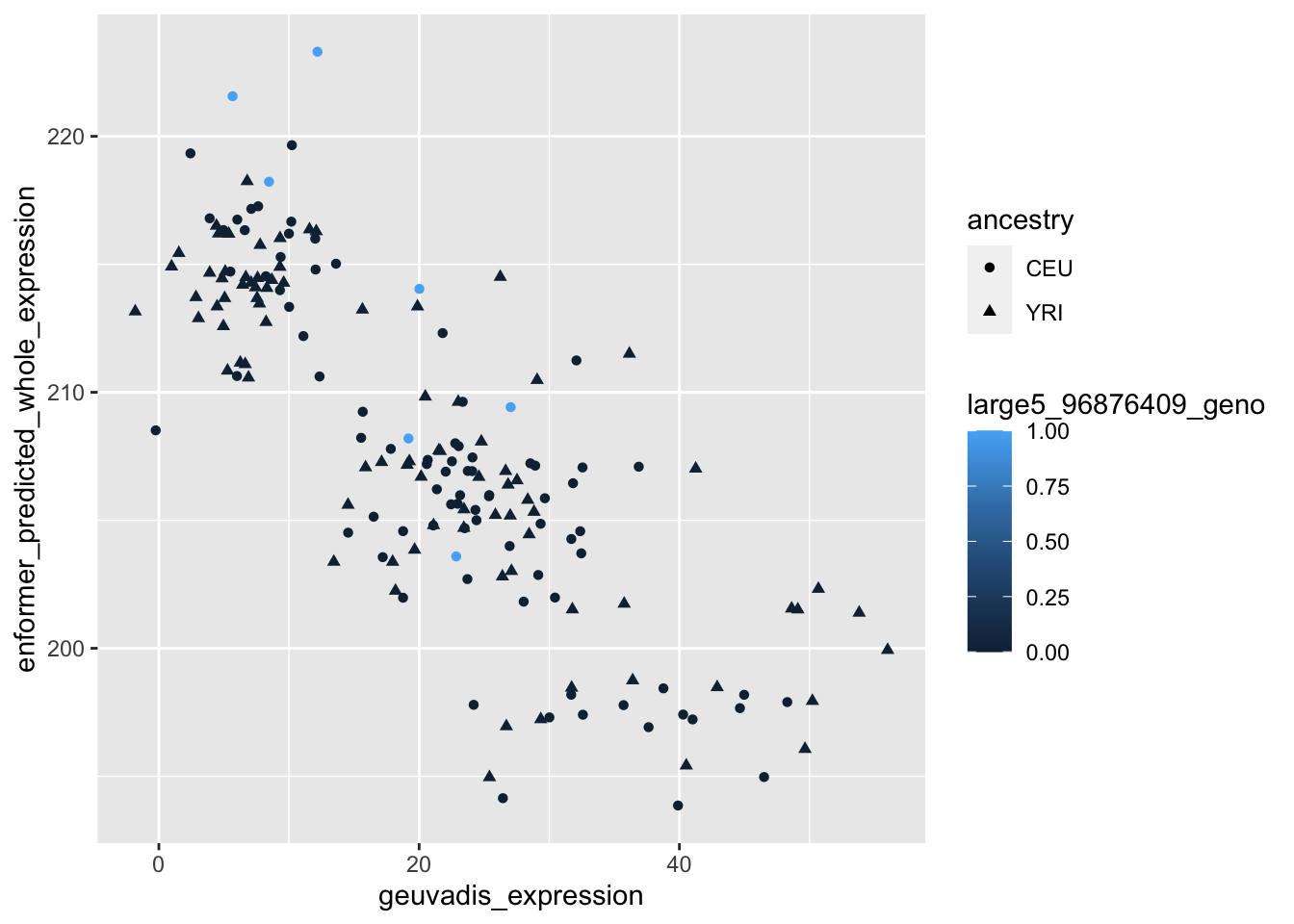
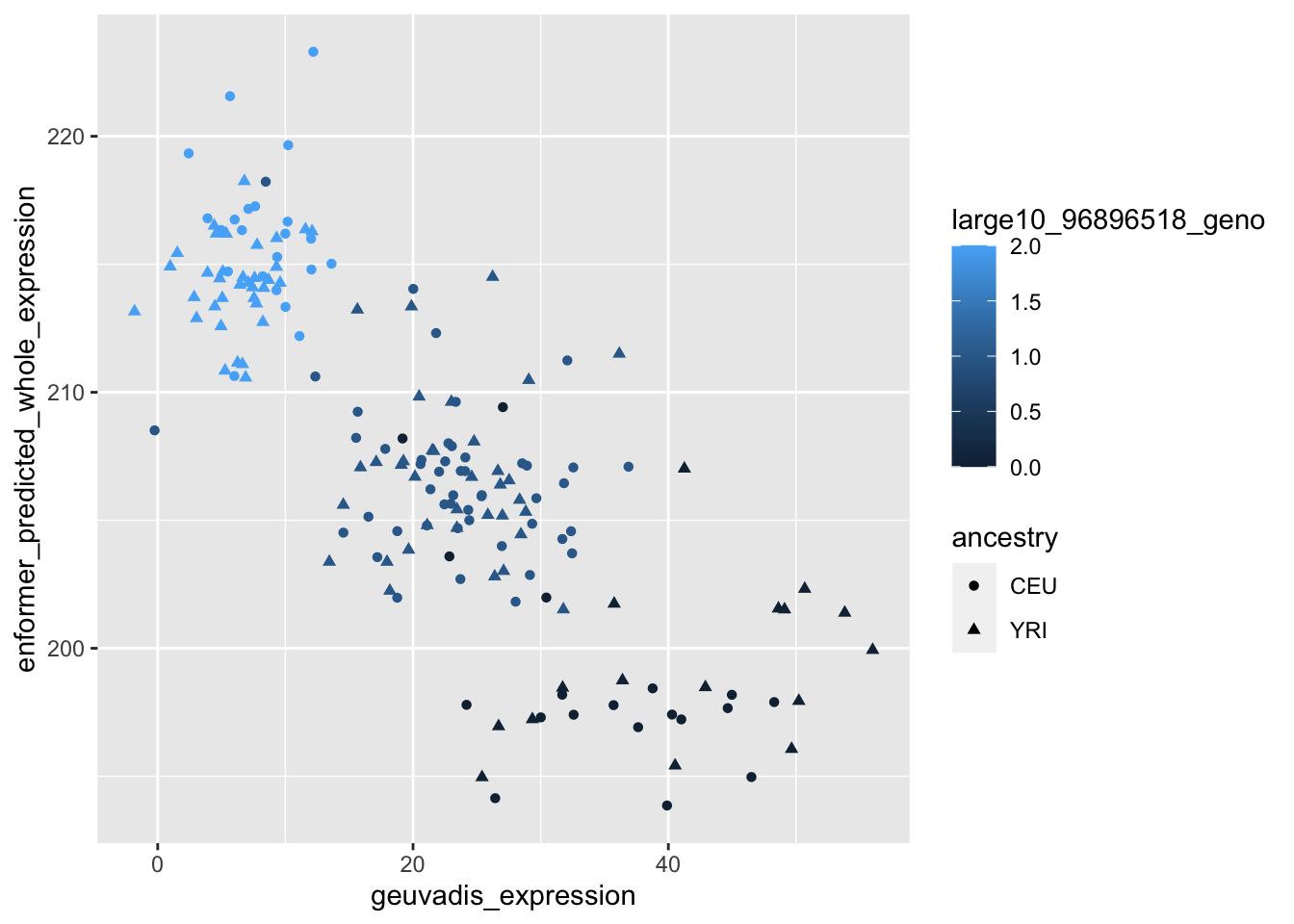
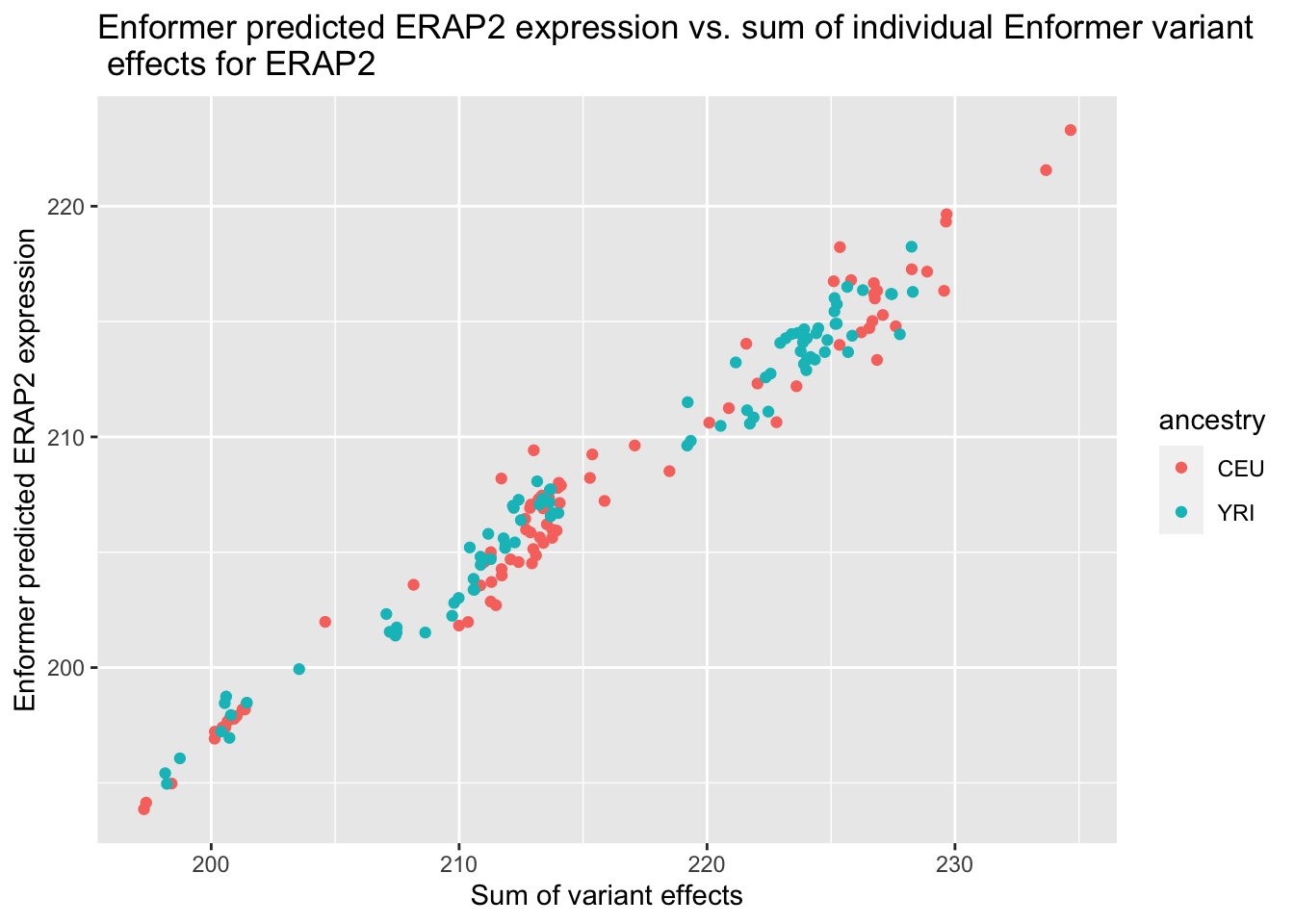
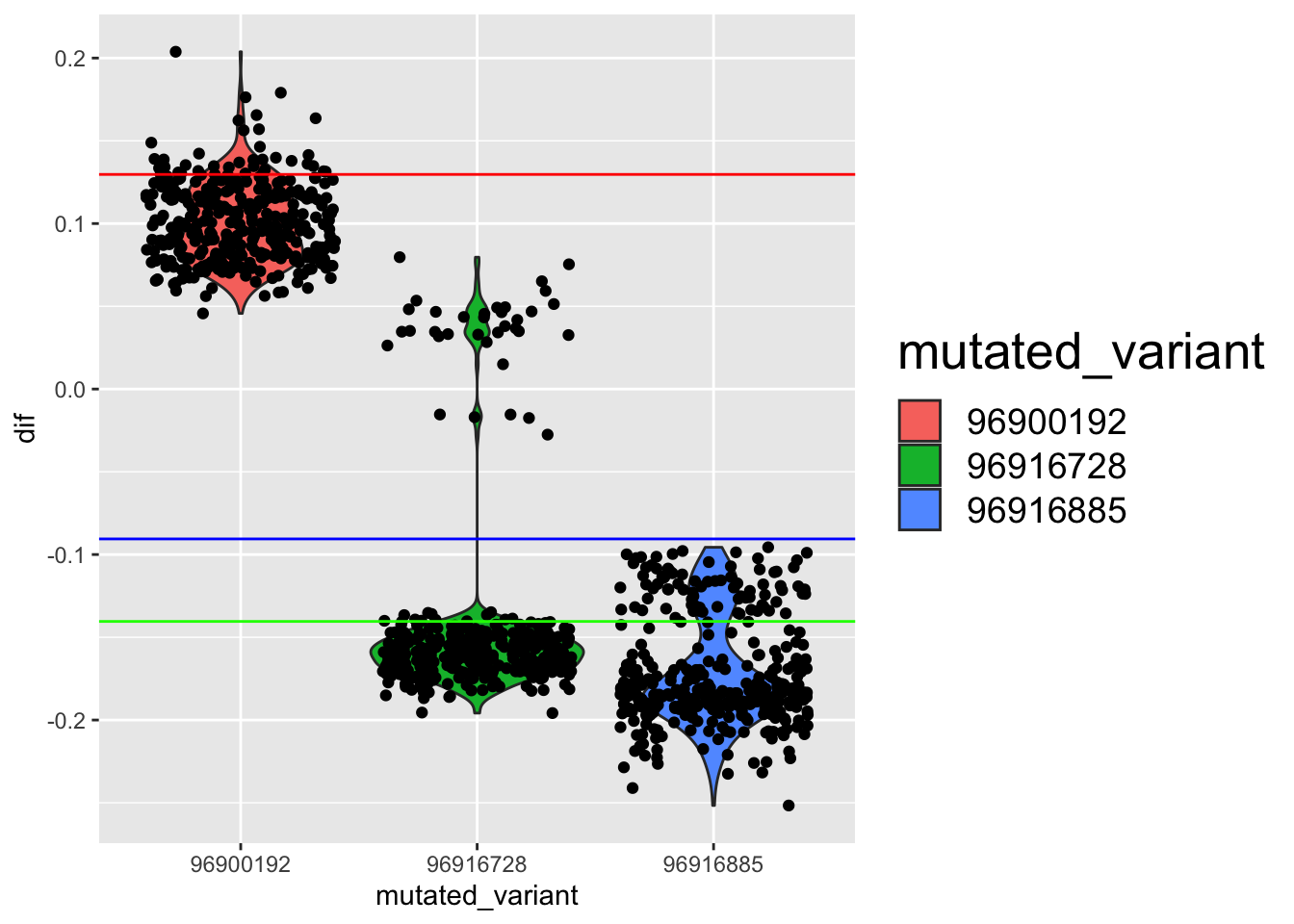
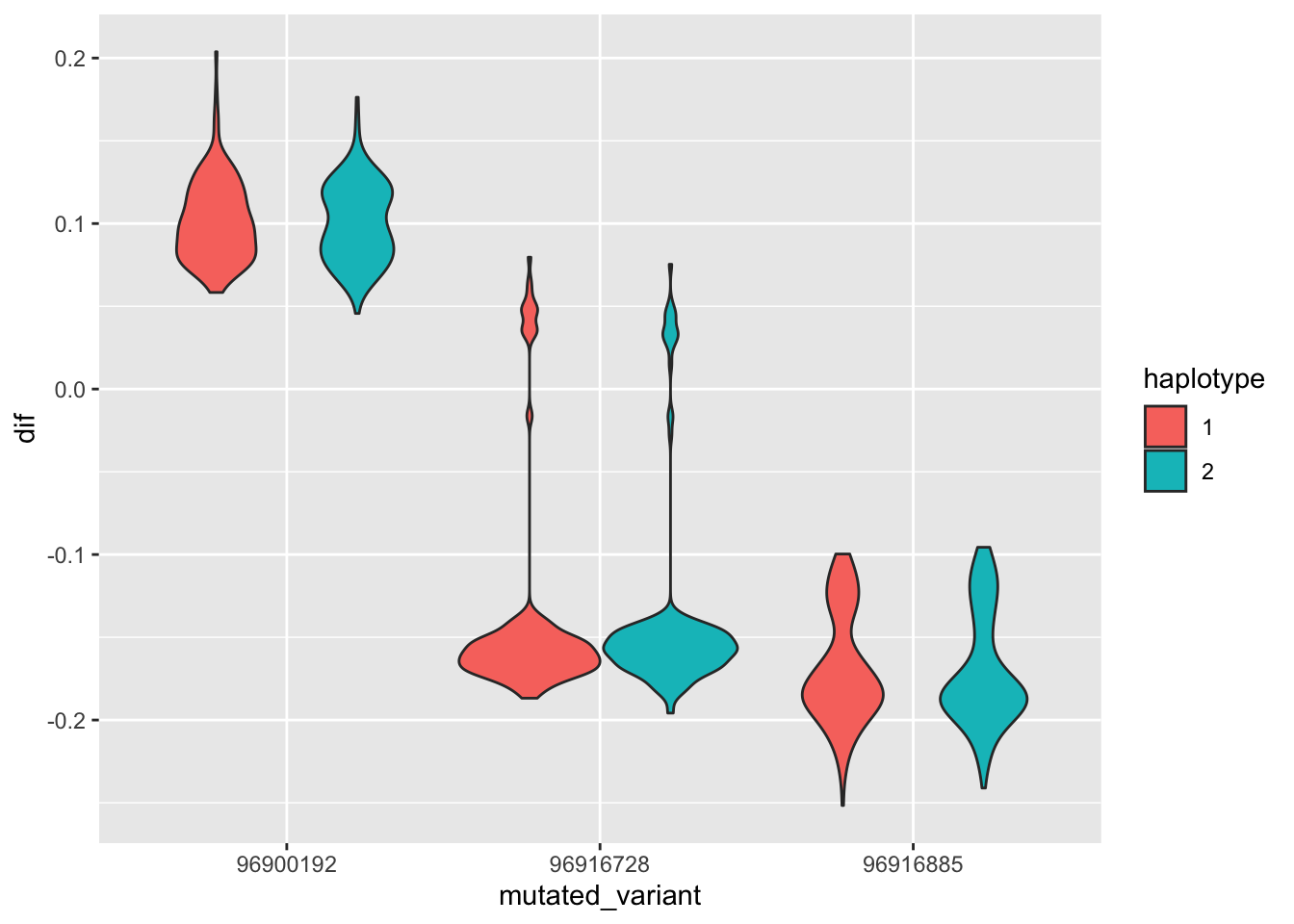
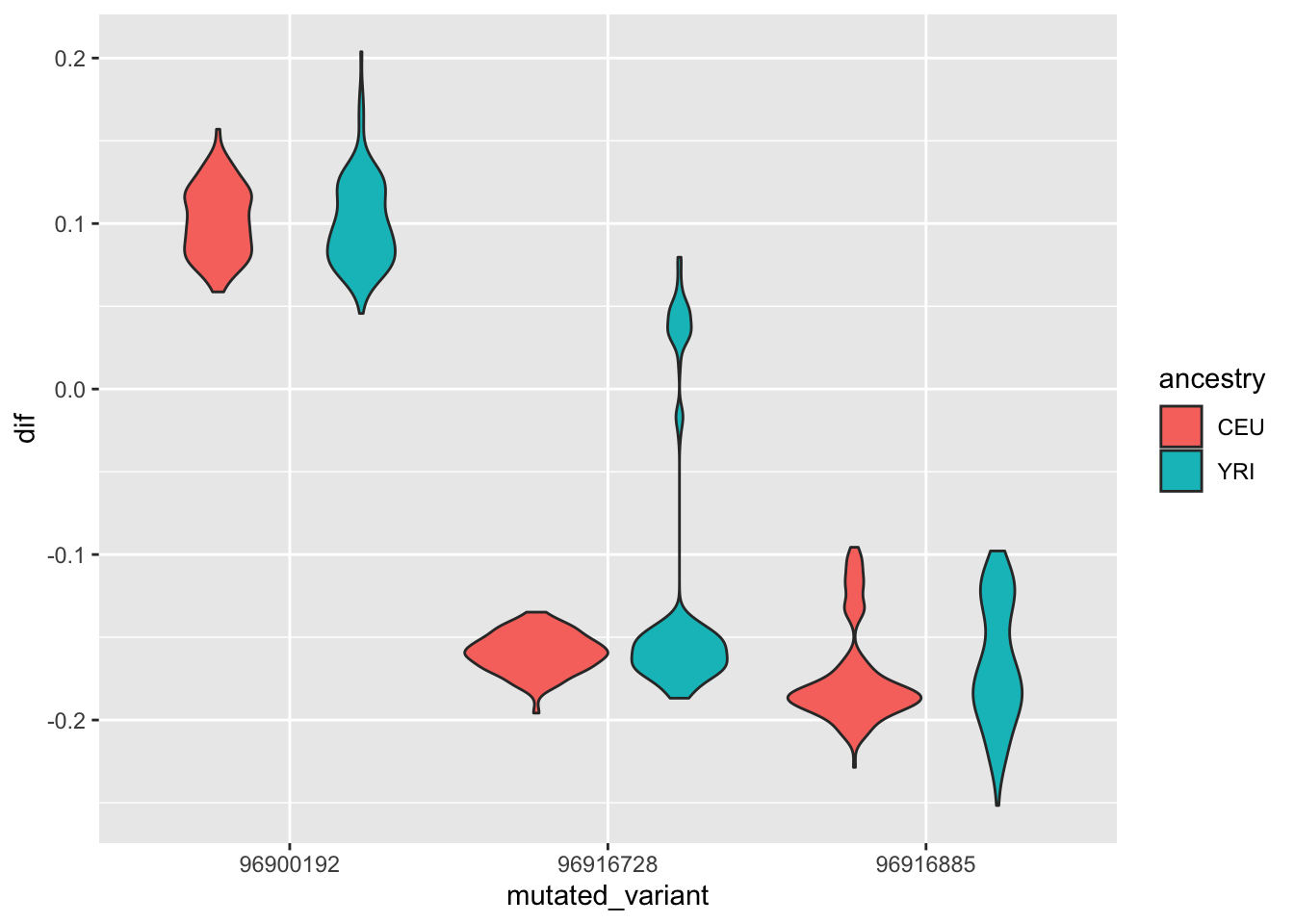
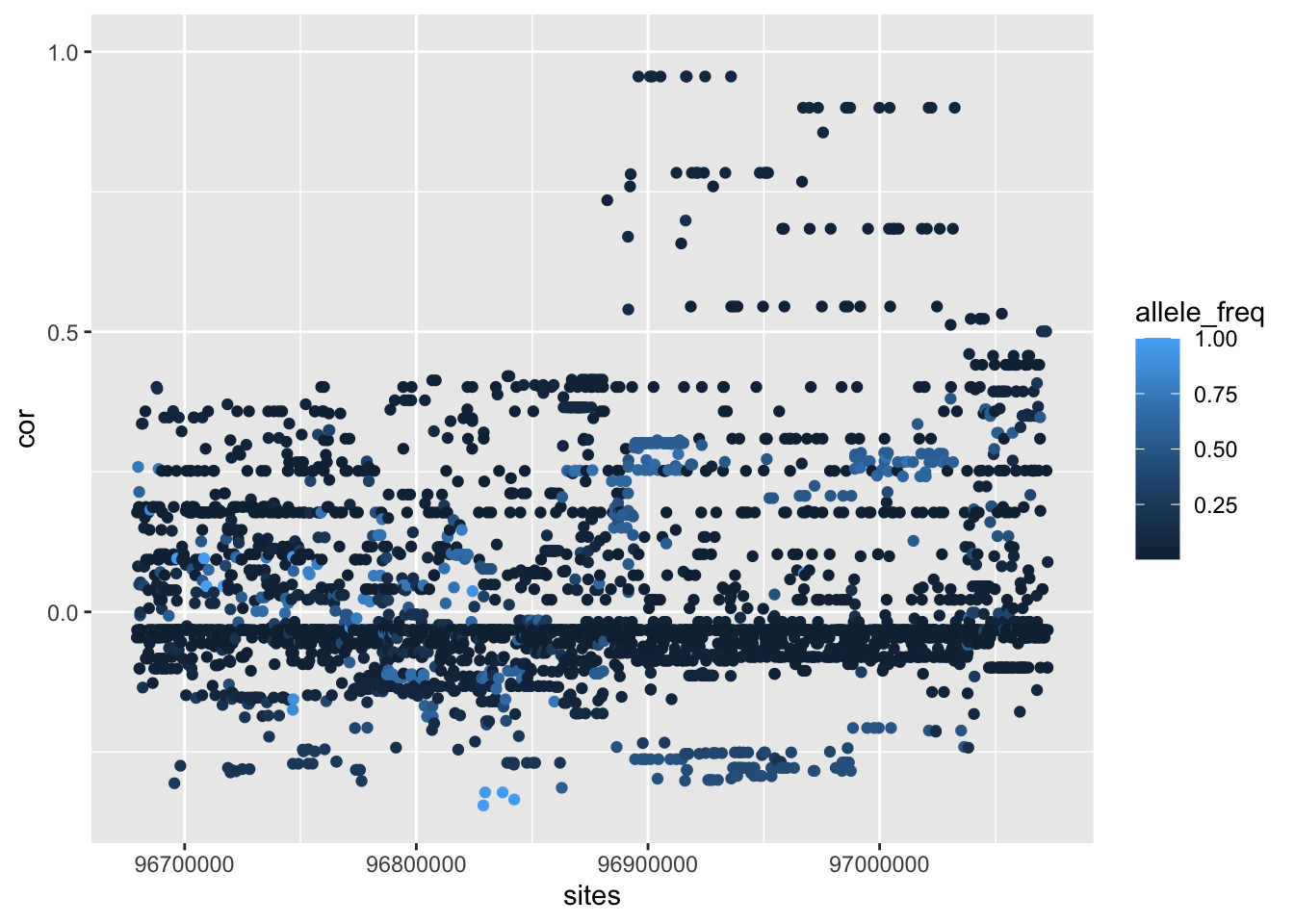
--- title: "analyzing-ERAP2-personalized-mutagenesis" author: "Saideep Gona" date: "2023-04-04" format: html: code-fold: true code-summary: "Show the code" execute: freeze: true warning: false --- ```{r} #| label: Set up box storage directory suppressMessages (library (tidyverse))suppressMessages (library (glue))= "/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/Daily-Blog-Sai" ## COPY THE DATE AND SLUG fields FROM THE HEADER = "analyzing-ERAP2-personalized-mutagenesis" ## copy the slug from the header = '2023-04-04' ## copy the date from the blog's header here = glue ("{PRE}/{bDATE}-{SLUG}" )if (! file.exists (DATA)) system (glue:: glue ("mkdir {DATA}" ))= DATA``` # Context ### Load in relevant metadata ```{r} = "/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/metadata" <- read_delim (file.path (metadata_dir,"relationships_w_pops.txt" ))rownames (pop_metadata) <- pop_metadata$ IID<- read_csv (file.path (metadata_dir,"ref_local_ancestry" ,"gene_annotations.csv" ))<- as.vector (gene_annotations$ ...1 )<- gene_annotations[,2 : ncol (gene_annotations)]rownames (gene_annotations) <- genes"ENSG00000164308" ,]sum (rowSums (gene_annotations)== 0 )hist (rowSums (gene_annotations))# Geuvadis expression <- "/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/enformer_geuvadis/enformer_portability_analysis" <- read_delim (file.path (dset_dir,"GD462.GeneQuantRPKM.50FN.samplename.resk10.txt" ))<- sapply (str_split (geuvadis_full$ Gene_Symbol, " \\ ." ),` [ ` ,1 )<- as.data.frame (geuvadis_full[,5 : ncol (geuvadis_full)])rownames (geuvadis) <- geuvadis_genes_symbols``` ```{r} = "/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/enformer_geuvadis/ERAP2_personalized_mutagenesis" <- read_csv (file.path (data_dir,"genotypes_df.csv" ))<- as.vector (genotype_data[,1 ])[[1 ]]rownames (genotype_data) <- as.vector (genotype_data[,1 ])[[1 ]]1 ] <- NULL rownames (genotype_data) <- variant_ids<- ifelse (genotype_data== "1|1" ,2 ,ifelse (genotype_data== "0|0" ,0 ,1 ))rownames (allele_count_df) <- variant_ids<- data.frame (af= rowSums (allele_count_df)/ (2 * ncol (allele_count_df* 2 )))``` ## Effect size of reference variants alone ```{r} = "/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/enformer_geuvadis/ERAP2_personalized_mutagenesis" <- read_csv (file.path (data_dir,"erap2_personalized_LCL_REF_quantification_haplo1.csv" ))<- haplo1_ref[,3 : ncol (haplo1_ref)]<- read_csv (file.path (data_dir,"erap2_personalized_LCL_REF_quantification_haplo1_rc.csv" ))<- haplo1_ref_rc[,3 : ncol (haplo1_ref_rc)]= data.frame (t ((haplo1_ref_rc + haplo1_ref)/ 2 ))colnames (haplo1_ref_ave) <- c ("expression" )<- as.numeric (haplo1_ref_ave[1 ,1 ])<- read_csv (file.path (data_dir,"erap2_personalized_LCL_REF_quantification_haplo2.csv" ))<- haplo2_ref[,3 : ncol (haplo2_ref)]<- read_csv (file.path (data_dir,"erap2_personalized_LCL_REF_quantification_haplo2_rc.csv" ))<- haplo2_ref_rc[,3 : ncol (haplo2_ref_rc)]= data.frame (t ((haplo2_ref_rc + haplo2_ref)/ 2 ))colnames (haplo2_ref_ave) <- c ("expression" )``` ```{r} <- haplo2_ref_ave - haplo1_ref_avecolnames (ref_dif) <- c ("dif" )$ ids <- rownames (ref_dif)<- ref_dif %>% separate (ids,into = c ("individual" ,"mutated_variant" ,"ref" ))$ pos <- as.numeric (ref_dif$ mutated_variant)$ voi <- ifelse (ref_dif$ mutated_variant %in% c ("96916885" ,"96916728" ,"96900192" ),TRUE ,FALSE )$ voi_sizes <- as.numeric (ifelse (ref_dif$ mutated_variant %in% c ("96916885" ,"96916728" ,"96900192" ),15 ,1 ))$ allele_freq <- as.numeric (variant_metadata_df[ref_dif$ mutated_variant,1 ])``` ````{r} ggplot (ref_dif) + geom_point (aes (x= pos,y= dif, color = allele_freq), alpha= 0.5 ) + scale_x_continuous (breaks = seq (96875986-80000 , 96875986 + 80000 , by = 20000 )) + theme (axis.text.x = element_text (angle = 90 , vjust = 0.5 , hjust= 1 )) + geom_vline (xintercept = 96875986 ) + ylab ("variant effect magnitude" ) + xlab ("position on chr5" ) + ggtitle ("In-silico mutagenesis of ERAP2 variants" )``` ```{r} ggplot (ref_dif) + geom_point (aes (x= allele_freq, y= dif))``` Ok, let's see how well some of these interesting variants correlate with the expression patterns we are seeing: Calculate true individual-specific expression estimates, this first requires loading the personalized expression: ```{r} library (tidyverse)<- read_csv (file.path (data_dir,"erap2_personalized_LCL_quantification_haplo1.csv" ))<- haplo1[,3 : ncol (haplo1)]<- read_csv (file.path (data_dir,"erap2_personalized_LCL_quantification_haplo1_rc.csv" ))<- haplo1_rc[,3 : ncol (haplo1_rc)]= data.frame (t ((haplo1_rc + haplo1)/ 2 ))colnames (haplo1_ave) <- c ("expression" )<- read_csv (file.path (data_dir,"erap2_personalized_LCL_quantification_haplo2.csv" ))<- haplo2[,3 : ncol (haplo2)]<- read_csv (file.path (data_dir,"erap2_personalized_LCL_quantification_haplo2_rc.csv" ))<- haplo2_rc[,3 : ncol (haplo2_rc)]= data.frame (t ((haplo2_rc + haplo2)/ 2 ))colnames (haplo2_ave) <- c ("expression" )``` ```{r} $ ids <- rownames (haplo1_ave)<- haplo1_ave %>% separate (ids,into = c ("individual" ,"mutated_variant" ,"mutated_genotype" ))$ haplotype <- "1" <- haplo1_ave[haplo1_ave$ mutated_genotype == "REF" ,]<- haplo1_ave[haplo1_ave$ mutated_genotype == "ALT" ,]$ ids <- rownames (haplo2_ave)<- haplo2_ave %>% separate (ids,into = c ("individual" ,"mutated_variant" ,"mutated_genotype" ))$ haplotype <- "2" <- haplo2_ave[haplo2_ave$ mutated_genotype == "REF" ,]<- haplo2_ave[haplo2_ave$ mutated_genotype == "ALT" ,]<- rbind (haplo1_ref,haplo2_ref)<- rbind (haplo1_alt,haplo2_alt)<- data.frame (individual= ref_stack$ individual,haplotype= ref_stack$ haplotype,mutated_variant= ref_stack$ mutated_variant, ref_expression= ref_stack$ expression, alt_expression= alt_stack$ expression)``` ```{r} <- c ()for (ind in colnames (genotype_data)) {= 0 = str_split (genotype_data["96916885" ,ind], "|" )if (cur_geno[[1 ]][2 ] == "0" ) {= paste (ind,"96916885" ,"REF" , sep= "_" )= expression + haplo1_ave[id_tag,]$ expressionelse {= paste (ind,"96916885" ,"ALT" , sep= "_" )= expression + haplo1_ave[id_tag,]$ expressionif (cur_geno[[1 ]][4 ] == "0" ) {= paste (ind,"96916885" ,"REF" , sep= "_" )= expression + haplo2_ave[id_tag,]$ expressionelse {= paste (ind,"96916885" ,"ALT" , sep= "_" )= expression + haplo2_ave[id_tag,]$ expression<- c (erap2_expression, expression)``` Let's also grab the large effect size variants from the reference mutagenesis: ```{r} $ dif > 4 ,]``` ```` {r} erap2_expression_df <- data.frame(enformer_predicted_whole_expression=erap2_expression, geuvadis_expression = as.numeric(geuvadis["ENSG00000164308",colnames(genotype_data)]), fm_96916885_geno=allele_count_df["96916885",], splice_96900192_geno=allele_count_df["96900192",], fm_96916728_geno=allele_count_df["96916728",], large5_96876409_geno=allele_count_df["96876409",], large10_96896518_geno=allele_count_df["96896518",]) rownames(erap2_expression_df) <- colnames(genotype_data) erap2_expression_df$ancestry <- as.vector(pop_metadata[rownames(erap2_expression_df),]$population) ggplot(erap2_expression_df) + geom_point(aes(x=geuvadis_expression, y=enformer_predicted_whole_expression, color=ancestry)) + xlab("Geuvadis Expression for ERAP2") + ylab("Enformer predicted ERAP2 expression") + ggtitle("Predicted vs. Observed ERAP2 expression across individuals") ggplot(erap2_expression_df) + geom_point(aes(x=geuvadis_expression, y=enformer_predicted_whole_expression, color=splice_96900192_geno, shape=ancestry)) ggplot(erap2_expression_df) + geom_point(aes(x=geuvadis_expression, y=enformer_predicted_whole_expression, color=fm_96916885_geno, shape=ancestry)) ggplot(erap2_expression_df) + geom_point(aes(x=geuvadis_expression, y=enformer_predicted_whole_expression, color=fm_96916728_geno, shape=ancestry)) ggplot(erap2_expression_df) + geom_point(aes(x=geuvadis_expression, y=enformer_predicted_whole_expression, color=large5_96876409_geno, shape=ancestry)) ggplot(erap2_expression_df) + geom_point(aes(x=geuvadis_expression, y=enformer_predicted_whole_expression, color=large10_96896518_geno, shape=ancestry)) ``` 's generate predicted personalized gene expression by summing up individual variant effects with the reference prediction as a baseline: ```{r} allele_count_mat <- as.data.frame(allele_count_df) dif_mat <- data.frame(difs = ref_dif$dif) rownames(dif_mat) <- ref_dif$mutated_variant allele_count_mat <- t(as.matrix(allele_count_mat[rownames(dif_mat),])) dif_mat <- as.matrix(dif_mat) linear_predictions <- allele_count_mat %*% dif_mat + reference_hap_expression*2 erap2_expression_df$linear_pred <- as.vector(linear_predictions) ggplot(erap2_expression_df) + geom_point(aes(x=linear_predictions, y= enformer_predicted_whole_expression, color=ancestry)) + ylab("Enformer predicted ERAP2 expression") + xlab("Sum of variant effects") + ggtitle("Enformer predicted ERAP2 expression vs. sum of individual Enformer variant \n effects for ERAP2") ``` The above plot plots the individualized expression predictions Clearly, ERAP2 is well predicted as a linear sum of individual variant effects. This supports the conclusion that the large effect variant is at play here. ## personalized mutagenesis After loading in the personalized expression estimates We can also annotate each value with data for the individual, haplotype, mutated variant, and genotype of mutated variant. ```{r} library(tidyverse) haplo1 <- read_csv(file.path(data_dir,"erap2_personalized_LCL_quantification_haplo1.csv")) haplo1 <- haplo1[,3:ncol(haplo1)] haplo1_rc <- read_csv(file.path(data_dir,"erap2_personalized_LCL_quantification_haplo1_rc.csv")) haplo1_rc<- haplo1_rc[,3:ncol(haplo1_rc)] haplo1_ave = data.frame(t((haplo1_rc + haplo1)/2)) colnames(haplo1_ave) <- c("expression") haplo2 <- read_csv(file.path(data_dir,"erap2_personalized_LCL_quantification_haplo2.csv")) haplo2 <- haplo2[,3:ncol(haplo2)] haplo2_rc <- read_csv(file.path(data_dir,"erap2_personalized_LCL_quantification_haplo2_rc.csv")) haplo2_rc <- haplo2_rc[,3:ncol(haplo2_rc)] haplo2_ave = data.frame(t((haplo2_rc + haplo2)/2)) colnames(haplo2_ave) <- c("expression") ``` ```{r} haplo1_ave$ids <- rownames(haplo1_ave) haplo1_ave <- haplo1_ave %>% separate(ids,into = c("individual","mutated_variant","mutated_genotype")) haplo1_ave$haplotype <- "1" haplo1_ref <- haplo1_ave[haplo1_ave$mutated_genotype == "REF",] haplo1_alt <- haplo1_ave[haplo1_ave$mutated_genotype == "ALT",] haplo2_ave$ids <- rownames(haplo2_ave) haplo2_ave <- haplo2_ave %>% separate(ids,into = c("individual","mutated_variant","mutated_genotype")) haplo2_ave$haplotype <- "2" haplo2_ref <- haplo2_ave[haplo2_ave$mutated_genotype == "REF",] haplo2_alt <- haplo2_ave[haplo2_ave$mutated_genotype == "ALT",] ref_stack <- rbind(haplo1_ref,haplo2_ref) alt_stack <- rbind(haplo1_alt,haplo2_alt) full_df <- data.frame(individual=ref_stack$individual,haplotype=ref_stack$haplotype,mutated_variant=ref_stack$mutated_variant, ref_expression=ref_stack$expression, alt_expression=alt_stack$expression) ``` We now have collected all our estimates into a concise data frame. Next we can calculate some effect-size related estimates by comparing the alt,ref ```{r} full_df$dif <- full_df$alt_expression-full_df$ref_expression full_df$fc <- full_df$alt_expression/full_df$ref_expression full_df$logfc <- log2(full_df$fc) full_df$log_ref <- log(full_df$ref_expression) full_df$log_alt <- log(full_df$alt_expression) ``` We can also annotate our results by individual ancestry ```{r} full_df$ancestry <- as.vector(pop_metadata[full_df$individual,]$population) ``` ```{r} # TODO Plot against allele frequency # TODO Color by ancestry ggplot(full_df) + geom_violin(aes(x=mutated_variant, y=dif, fill=mutated_variant)) + geom_jitter(aes(x=mutated_variant, y=dif))+geom_hline(yintercept = ref_dif[ref_dif$mutated_variant=="96916885","dif"], color="blue") + geom_hline(yintercept = ref_dif[ref_dif$mutated_variant=="96916728","dif"], color="green") + geom_hline(yintercept = ref_dif[ref_dif$mutated_variant=="96900192","dif"], color="red") +scale_color_manual(name=' Personalized vs. ref mutagenesis', breaks=c(' 96916885 ', ' 96916728 ', ' 96900192 '), values=c(' 96916885 '=' red', ' 96916728 '=' blue', ' 96900192 '=' green')) + theme(legend.title=element_text(size=20), legend.text=element_text(size=14)) ggplot(full_df) + geom_violin(aes(x=mutated_variant, y=dif, fill=haplotype)) ggplot(full_df) + geom_violin(aes(x=mutated_variant, y=dif, fill=ancestry)) ``` The distributions are certainly interesting. It would be good to try and identify if the 96916728 "positive" group is an interaction with a specific variant present more frequently in that subgroup. ```{r} full_df_fm_96916728_pos <- full_df[(full_df$dif > 0) & (full_df$mutated_variant == 96916728),] fm_96916728_pos_individuals <- unique(full_df_fm_96916728_pos$individual) allele_count_sub <- as.data.frame(t(allele_count_df[rownames(dif_mat),])) allele_count_sub$fm_96916728_pos <- rownames(allele_count_sub) %in% fm_96916728_pos_individuals fm_96916728_pos_cor <- as.data.frame(cor(allele_count_sub[,colnames(allele_count_sub != "fm_96916728_pos")],allele_count_sub$fm_96916728_pos)) fm_96916728_pos_cor$sites <- as.numeric(rownames(fm_96916728_pos_cor)) colnames(fm_96916728_pos_cor) <- c("cor", "sites") fm_96916728_pos_cor$allele_freq <- as.numeric(variant_metadata_df[rownames(fm_96916728_pos_cor),1]) ggplot(fm_96916728_pos_cor) + geom_point(aes(x=sites,y=cor, col=allele_freq)) ```