suppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="correcting-antisense-gene-prediction"## copy the slug from the headerbDATE='2023-04-28'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA
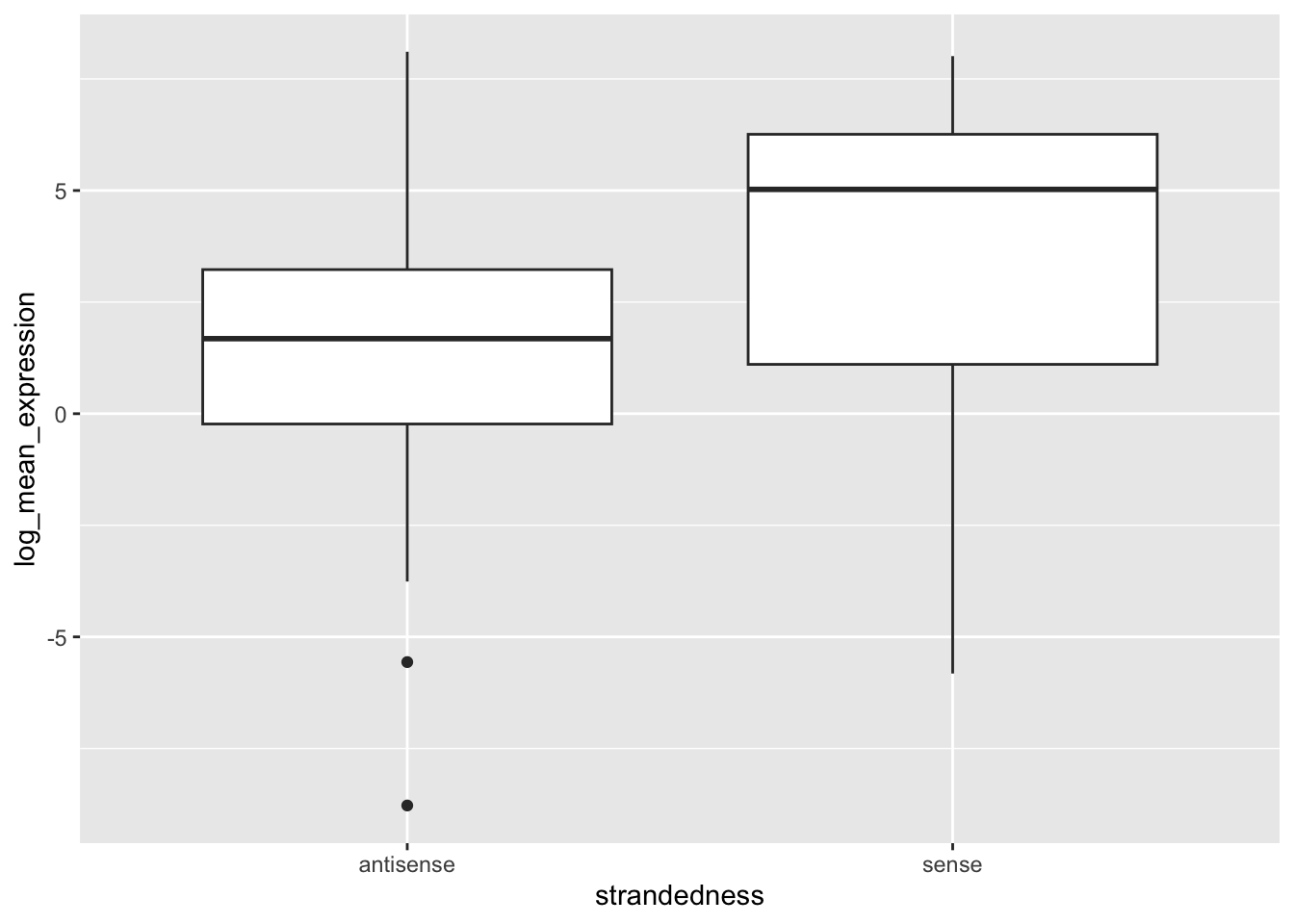
Ok so clearly the antisense genes were being miscalculated, although from prior analysis there doesn’t seem to be a major deviation in terms of the direction of the correlations with geuvadis predictions.
To fix this, two things need to be done:
1.) TSS sites need to be corrected 2.) Centering of antisense genes should be done on the end of the gene body rather than the beginning
With regards to 1.), we have decided to use BioMart for extraction for a few reasons. First of all, there is a lesser chance of making these types of mistakes with manual parsing of annotation files. Second, they specifically list the canonical transcript for each gene, which is likely the best location to center the sequence windows on. It also essentially solves 2.).
As such it is a good time to update my gene metadata files using the BioMart data. This is below:
---title: "correcting-antisense-gene-prediction"author: "Saideep Gona"date: "2023-04-28"format: html: code-fold: true code-summary: "Show the code"execute: freeze: true warning: false---```{r}#| label: Set up box storage directorysuppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="correcting-antisense-gene-prediction"## copy the slug from the headerbDATE='2023-04-28'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA``````{r}library(tidyverse)dset_dir <-file.path("/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/enformer_geuvadis/enformer_epistasis")haplo1 <-as.data.frame(read_csv(file.path(dset_dir,"CAGE_lcl_enformer_geuvadis_summed_haplo1_filtered.csv")))rownames(haplo1) <- haplo1$...1haplo1$...1<-NULLmetadata_dir ="/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/metadata"gene_metadata <-as.data.frame(read_delim(file.path(metadata_dir,"gene_metadata_full.csv")))rownames(gene_metadata) <-as.character(gene_metadata$...1)gene_metadata$...1<-NULLhaplo1 <- haplo1[rownames(haplo1) %in%rownames(gene_metadata),]gene_metadata <- gene_metadata[rownames(gene_metadata) %in%rownames(haplo1),]gene_metadata <- gene_metadata[rownames(haplo1),]haplo1_sense <- haplo1[gene_metadata$strand =="+",]haplo1_antisense <- haplo1[gene_metadata$strand =="-",]```There are 9618 sense (+) genes and 9408 antisense (-) genes in the cohort```{r}sense_means <-log(rowMeans(haplo1_sense))antisense_means <-log(rowMeans(haplo1_antisense))plot_sense_df <-data.frame(log_mean_expression=c(sense_means,antisense_means), strandedness=c(rep("sense",length(sense_means)),rep("antisense",length(antisense_means))))ggplot(plot_sense_df)+geom_boxplot(aes(x=strandedness,y=log_mean_expression))```Ok so clearly the antisense genes were being miscalculated, although from prior analysis there doesn't seem to be a major deviation in terms of the direction of the correlations with geuvadis predictions. To fix this, two things need to be done:1.) TSS sites need to be corrected2.) Centering of antisense genes should be done on the end of the gene body rather than the beginning With regards to 1.), we have decided to use BioMart for extraction for a few reasons. First of all, there is a lesser chance of making these types of mistakes with manual parsing of annotation files. Second, they specifically list the canonical transcript for each gene, which is likely the best location to center the sequence windows on. It also essentially solves 2.).As such it is a good time to update my gene metadata files using the BioMart data. This is below:```{r}library(biomaRt)mart <-useMart(biomart ="ENSEMBL_MART_ENSEMBL", dataset ="hsapiens_gene_ensembl", host ='www.ensembl.org')protein_coding_TSS <- biomaRt::getBM(attributes =c("ensembl_gene_id","external_gene_name", "chromosome_name","transcript_biotype", "transcript_start", "transcript_end", "transcription_start_site", "transcript_is_canonical", "transcript_count"), filters =c("transcript_biotype","chromosome_name"),values =list("protein_coding",c(1:22, 'X', 'Y')), mart = mart)```we can reformat this to something more familiar:```{r}canonical_TSS <- protein_coding_TSS %>%filter(protein_coding_TSS$transcript_is_canonical ==1)canonical_TSS$target_regions <-paste0("chr",canonical_TSS$chromosome_name,"_",canonical_TSS$transcription_start_site,"_",canonical_TSS$transcription_start_site+2)# write_csv(canonical_TSS$target_regions "")```