suppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="testing_ancestry_differences"## copy the slug from the headerbDATE='2023-05-30'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA
Context
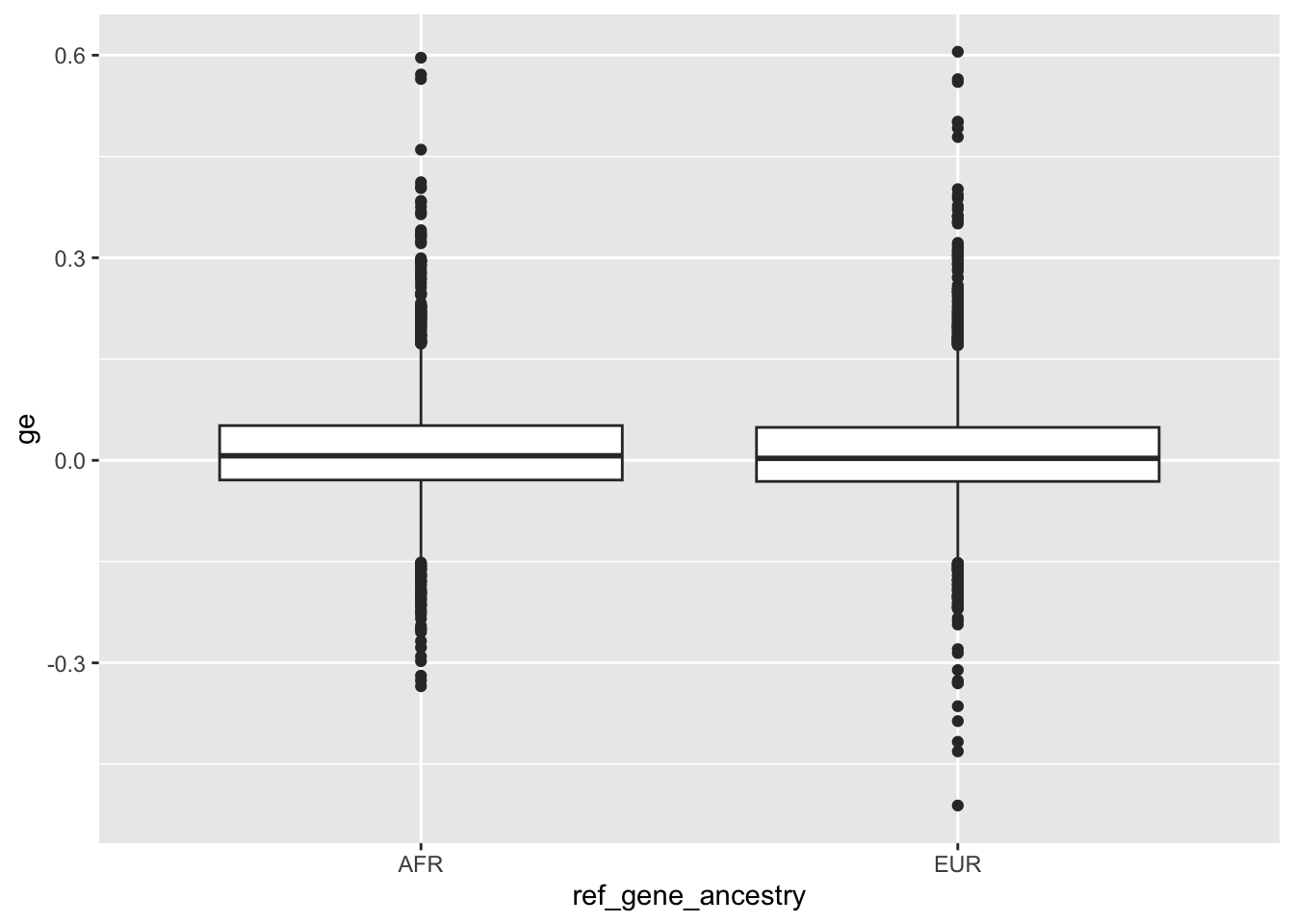
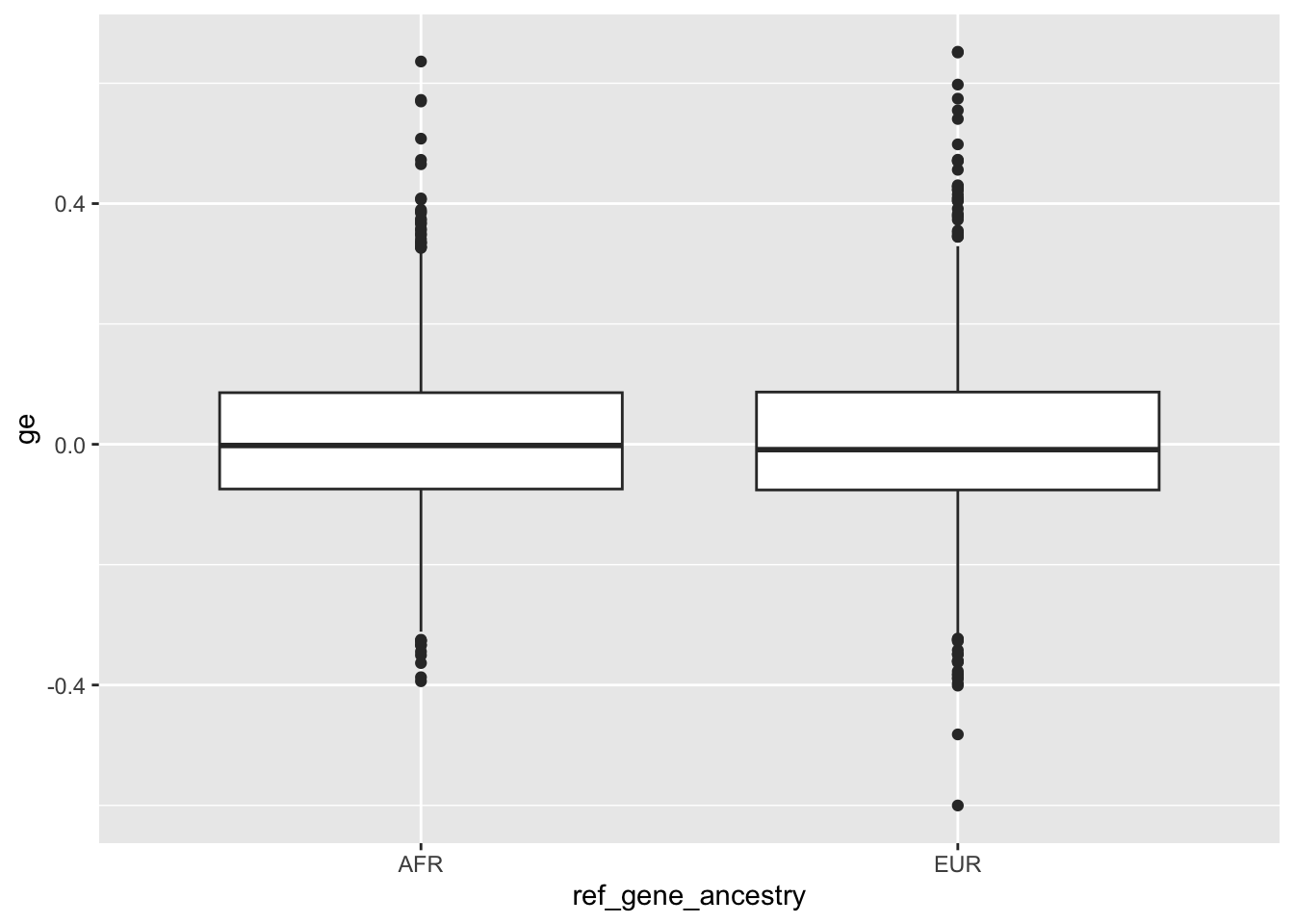
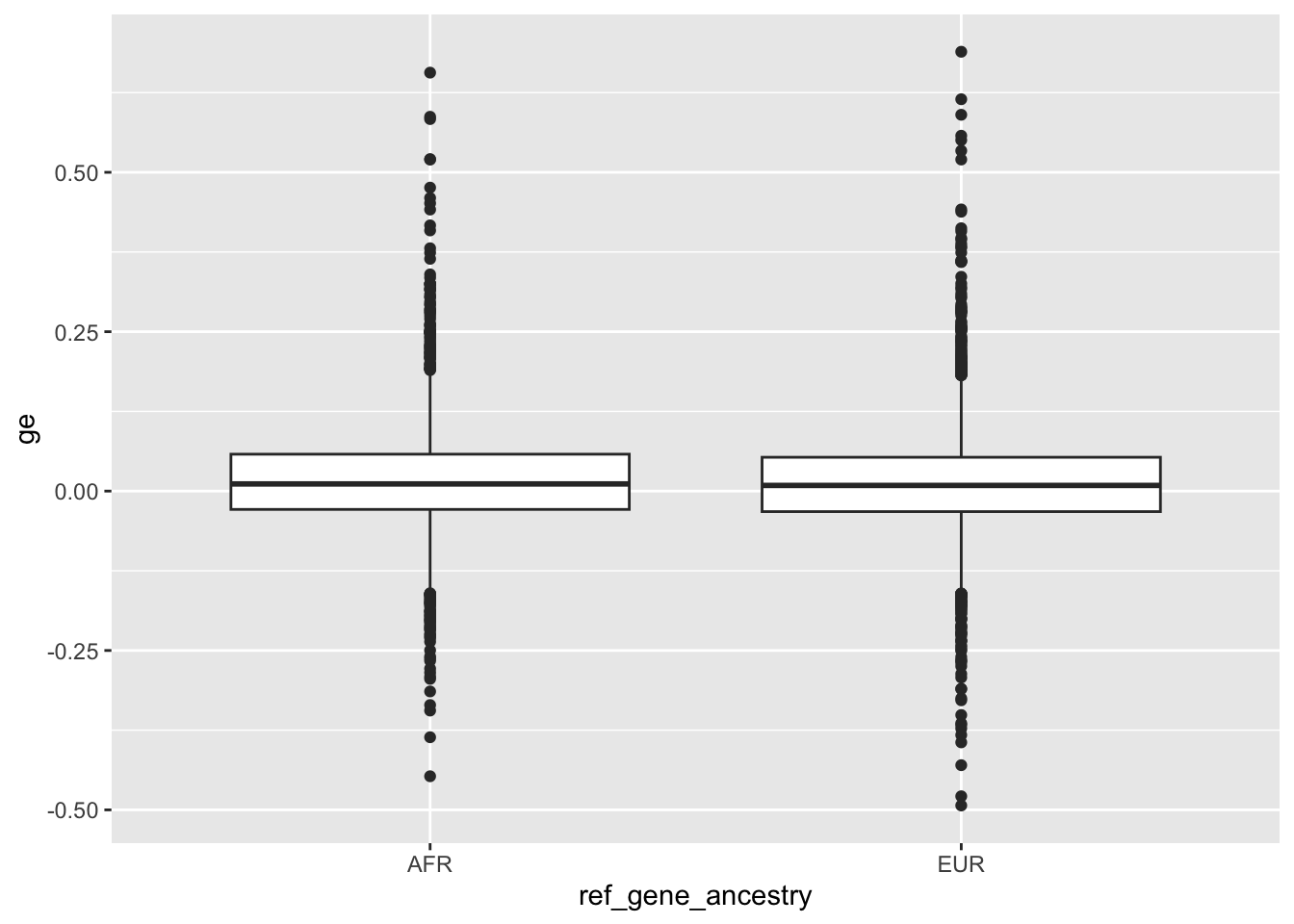
Previously I have collected local ancestry information for different genes (03-27). The hypothesis here is that the mismatches observed between local ancestry in the training reference genome, local ancestry in the Basenji targets (for example the lymphoblastoid cell line gm12878), and local ancestry in the inference sequence all contribute to issues observed with personalized prediction.
I didn’t fully test this hypothesis in enformer portability analysis notebook. Here I am going to more formally test it by collecting the following per-gene metrics:
1.) Local reference genome ancestry 2.) Average alternate (non-reference) allelic density per SNP in gene window from GM12878 lymphoblastoid cell line 3.) Direction of expression prediction correlation across individuals 4.) Potentially whether there is differential expression prediction between ancestry for the gene
We can update our gene metadata table with this added information once we have it.
Call:
glm(formula = AFR ~ ge, family = "binomial", data = corrs_df_plus_AFR_EUR)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.306 -1.168 -1.121 1.187 1.265
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.02543 0.02720 -0.935 0.350
ge 0.53248 0.32628 1.632 0.103
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7671.3 on 5533 degrees of freedom
Residual deviance: 7668.6 on 5532 degrees of freedom
AIC: 7672.6
Number of Fisher Scoring iterations: 3
summary(glm(AFR~ge, data=corrs_df_plus_AFR_EUR, family ="binomial"))
Call:
glm(formula = AFR ~ ge, family = "binomial", data = corrs_df_plus_AFR_EUR)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.196 -1.169 -1.157 1.186 1.202
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.01934 0.02691 -0.718 0.473
ge 0.09539 0.21383 0.446 0.656
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7671.3 on 5533 degrees of freedom
Residual deviance: 7671.1 on 5532 degrees of freedom
AIC: 7675.1
Number of Fisher Scoring iterations: 3
Call:
glm(formula = AFR ~ ge, family = "binomial", data = corrs_df_plus_AFR_EUR)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.301 -1.168 -1.122 1.186 1.276
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.02588 0.02730 -0.948 0.343
ge 0.45381 0.30051 1.510 0.131
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7671.3 on 5533 degrees of freedom
Residual deviance: 7669.0 on 5532 degrees of freedom
AIC: 7673
Number of Fisher Scoring iterations: 3
Source Code
---title: "testing_ancestry_differences"author: "Saideep Gona"date: "2023-05-30"format: html: code-fold: true code-summary: "Show the code"execute: freeze: true warning: false---```{r}#| label: Set up box storage directorysuppressMessages(library(tidyverse))suppressMessages(library(glue))PRE ="/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/Daily-Blog-Sai"## COPY THE DATE AND SLUG fields FROM THE HEADERSLUG="testing_ancestry_differences"## copy the slug from the headerbDATE='2023-05-30'## copy the date from the blog's header hereDATA =glue("{PRE}/{bDATE}-{SLUG}")if(!file.exists(DATA)) system(glue::glue("mkdir {DATA}"))WORK=DATA```# ContextPreviously I have collected local ancestry information for different genes (03-27). The hypothesis here is that the mismatches observed between local ancestry in the training reference genome, local ancestry in the Basenji targets (for example the lymphoblastoid cell line gm12878), and local ancestry in the inference sequence all contribute to issues observed with personalized prediction.I didn't fully test this hypothesis in enformer portability analysis notebook. Here I am going to more formally test it by collecting the following per-gene metrics:1.) Local reference genome ancestry 2.) Average alternate (non-reference) allelic density per SNP in gene window from GM12878 lymphoblastoid cell line 3.) Direction of expression prediction correlation across individuals 4.) Potentially whether there is differential expression prediction between ancestry for the geneWe can update our gene metadata table with this added information once we have it.### Computing metrics of local ancestryLet's load our gene metadata table:```{r}library(ggplot2)library(tidyverse)gene_metadata <-read_table("/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/metadata/ref_local_ancestry/gene_annotations.csv")```### Calculate correlation across individuals```{r}#| label: Load Datasetsdset_dir <-file.path("/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/enformer_geuvadis/enformer_portability_analysis")# Load GEUVADISgeuvadis_full <-read_delim(file.path(dset_dir,"GD462.GeneQuantRPKM.50FN.samplename.resk10.txt"))geuvadis_genes_symbols <-sapply(str_split(geuvadis_full$Gene_Symbol, "\\."),`[`,1)geuvadis <-as.data.frame(geuvadis_full[,5:ncol(geuvadis_full)])rownames(geuvadis) <- geuvadis_genes_symbols# Load enformerenformer_haplo1 <-as.data.frame(read_csv(file.path(dset_dir,"CAGE_lcl_enformer_geuvadis_summed_haplo1_filtered.csv")))rownames(enformer_haplo1) <- enformer_haplo1$...1enformer_haplo1$...1<-NULL# rownames(enformer_haplo1) <- enformer_haplo1$X# enformer_haplo1$X <- NULLenformer_haplo2 <-as.data.frame(read_csv(file.path(dset_dir,"CAGE_lcl_enformer_geuvadis_summed_haplo2_filtered.csv")))rownames(enformer_haplo2) <- enformer_haplo2$Xenformer_haplo2$...1<-NULLenf_genes_symbols <-sapply(str_split(rownames(enformer_haplo1), "\\."),`[`,1)enformer <- (enformer_haplo1 + enformer_haplo2)rownames(enformer) <- enf_genes_symbols``````{r}metadata_dir ="/Users/saideepgona/Library/CloudStorage/Box-Box/imlab-data/data-Github/analysis-Sai/metadata"individual_metadata <-read.delim(file.path(metadata_dir,"igsr_samples.tsv"))rownames(individual_metadata) <- individual_metadata$Sample.nameAFR_ind <- individual_metadata[individual_metadata$Superpopulation.code =="AFR",]$Sample.nameEUR_ind <- individual_metadata[individual_metadata$Superpopulation.code =="EUR",]$Sample.namegene_metadata <-read.csv2(file.path(metadata_dir,"gene_metadata_full.csv"),row.names =1)``````{r}ge_ind <-intersect(colnames(geuvadis), colnames(enformer))AFR_ge_ind <-intersect(ge_ind,AFR_ind)EUR_ge_ind <-intersect(ge_ind,EUR_ind)ge_genes <-intersect(rownames(geuvadis), rownames(enformer))geuvadis_compare <- geuvadis[ge_genes, ge_ind]geuvadis_AFR_compare <- geuvadis[ge_genes, AFR_ge_ind]geuvadis_EUR_compare <- geuvadis[ge_genes, EUR_ge_ind]enformer_compare <- enformer[ge_genes, ge_ind]enformer_AFR_compare <- enformer[ge_genes, AFR_ge_ind]enformer_EUR_compare <- enformer[ge_genes, EUR_ge_ind]gene_meta_compare <- gene_metadata[ge_genes,]print(paste0("Number of individuals: ", length(ge_ind)))print(paste0("Number of individuals AFR: ", length(AFR_ge_ind)))print(paste0("Number of individuals EUR: ", length(EUR_ge_ind)))ge_genes_plus <- ge_genesprint(paste0("Number of genes +: ", length(gene_meta_compare[gene_meta_compare$strand=="+",]$best_ancestry)))print(paste0("Number of genes +,AFR: ", length(gene_meta_compare[gene_meta_compare$best_ancestry=="AFR"&gene_meta_compare$strand=="+",]$best_ancestry)))print(paste0("Number of genes -,EUR: ", length(gene_meta_compare[gene_meta_compare$best_ancestry=="EUR"&gene_meta_compare$strand=="+",]$best_ancestry)))```Correlate across all individuals per gene between Geuvadis, Enformer```{r}corr_gene_across_inds <-function(df_1,df_2) { df_1 <-scale(df_1) df_2 <-scale(df_2) corrs <-sapply(seq.int(dim(df_1)[1]), function(i) cor(as.numeric(df_1[i,]), as.numeric(df_2[i,])))names(corrs) <-rownames(df_1)return(corrs)}ge_corrs <-corr_gene_across_inds(geuvadis_compare, enformer_compare)corrs_df <-data.frame(ge=ge_corrs, ge_abs =abs(ge_corrs),genes=ge_genes, gene_names=rownames(gene_meta_compare), gene_h2=gene_meta_compare$h2_WholeBlood_TS, high_h2 = gene_meta_compare$h2_WholeBlood_TS >0.1, strand = gene_meta_compare$strand, ref_gene_ancestry=gene_meta_compare$best_ancestry)corrs_df_plus <- corrs_df[corrs_df$strand =="+",]corrs_df_plus_AFR_EUR <- corrs_df[corrs_df$ref_gene_ancestry %in%c("AFR","EUR"),]corrs_df_plus_AFR_EUR$AFR <-as.factor(ifelse(corrs_df_plus_AFR_EUR$ref_gene_ancestry =="AFR",1,0))corrs_df_plus_AFR_EUR$corr_direction <-ifelse(corrs_df_plus_AFR_EUR$ge >0,"+","-")ggplot(corrs_df_plus_AFR_EUR) +geom_boxplot(aes(x=ref_gene_ancestry,y=ge))plot(table(corrs_df_plus_AFR_EUR$ref_gene_ancestry, corrs_df_plus_AFR_EUR$corr_direction))summary(glm(AFR~ge, data=corrs_df_plus_AFR_EUR, family="binomial"))```Correlate across only AFR individuals```{r}corr_gene_across_inds <-function(df_1,df_2) { df_1 <-scale(df_1) df_2 <-scale(df_2) corrs <-sapply(seq.int(dim(df_1)[1]), function(i) cor(as.numeric(df_1[i,]), as.numeric(df_2[i,])))names(corrs) <-rownames(df_1)return(corrs)}ge_corrs <-corr_gene_across_inds(geuvadis_AFR_compare, enformer_AFR_compare)corrs_df <-data.frame(ge=ge_corrs, ge_abs =abs(ge_corrs),genes=ge_genes, gene_names=rownames(gene_meta_compare), gene_h2=gene_meta_compare$h2_WholeBlood_TS, high_h2 = gene_meta_compare$h2_WholeBlood_TS >0.1, strand = gene_meta_compare$strand, ref_gene_ancestry=gene_meta_compare$best_ancestry)corrs_df_plus <- corrs_df[corrs_df$strand =="+",]corrs_df_plus_AFR_EUR <- corrs_df[corrs_df$ref_gene_ancestry %in%c("AFR","EUR"),]corrs_df_plus_AFR_EUR$AFR <-as.factor(ifelse(corrs_df_plus_AFR_EUR$ref_gene_ancestry =="AFR",1,0))corrs_df_plus_AFR_EUR$corr_direction <-ifelse(corrs_df_plus_AFR_EUR$ge >0,"+","-")ggplot(corrs_df_plus_AFR_EUR) +geom_boxplot(aes(x=ref_gene_ancestry,y=ge))plot(table(corrs_df_plus_AFR_EUR$ref_gene_ancestry, corrs_df_plus_AFR_EUR$corr_direction))summary(glm(AFR~ge, data=corrs_df_plus_AFR_EUR, family ="binomial"))```Correlate across only EUR individuals```{r}corr_gene_across_inds <-function(df_1,df_2) { df_1 <-scale(df_1) df_2 <-scale(df_2) corrs <-sapply(seq.int(dim(df_1)[1]), function(i) cor(as.numeric(df_1[i,]), as.numeric(df_2[i,])))names(corrs) <-rownames(df_1)return(corrs)}ge_corrs <-corr_gene_across_inds(geuvadis_EUR_compare, enformer_EUR_compare)corrs_df <-data.frame(ge=ge_corrs, ge_abs =abs(ge_corrs),genes=ge_genes, gene_names=rownames(gene_meta_compare), gene_h2=gene_meta_compare$h2_WholeBlood_TS, high_h2 = gene_meta_compare$h2_WholeBlood_TS >0.1, strand = gene_meta_compare$strand, ref_gene_ancestry=gene_meta_compare$best_ancestry)corrs_df_plus <- corrs_df[corrs_df$strand =="+",]corrs_df_plus_AFR_EUR <- corrs_df[corrs_df$ref_gene_ancestry %in%c("AFR","EUR"),]corrs_df_plus_AFR_EUR$AFR <-as.factor(ifelse(corrs_df_plus_AFR_EUR$ref_gene_ancestry =="AFR",1,0))corrs_df_plus_AFR_EUR$corr_direction <-ifelse(corrs_df_plus_AFR_EUR$ge >0,"+","-")ggplot(corrs_df_plus_AFR_EUR) +geom_boxplot(aes(x=ref_gene_ancestry,y=ge))# ggplot(data.frame(table(corrs_df_plus_AFR_EUR$ref_gene_ancestry, corrs_df_plus_AFR_EUR$corr_direction)), aes(x=Var1,y=Freq,fill=Var2, group=Var1)) + geom_bar(stat="identity", position="dodge")plot(table(corrs_df_plus_AFR_EUR$ref_gene_ancestry, corrs_df_plus_AFR_EUR$corr_direction))summary(glm(AFR~ge, data=corrs_df_plus_AFR_EUR, family="binomial"))```