Need some unified functions for analyzing SPrediXcan results.
Format should be a dictionary with the following tiers:
GWAS trait
Model type
Dataframe with SPrediXcan results
Specify inputs
Code
import os,sysimport mixbox as mxcolor_pal = {"conditions": {"Flu": (255, 0, 0),"NI": (0, 0, 255), },"models": {"EnPACT":(255, 255, 255),"Standard":(0,0,0),"GTeX_WB":(0,64,0) },"subsets": {"immune": (255, 165, 0) }}input_files = {"T2D":{"Flu_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/Flu_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto/intermediates/TWAS/T2D/T2D.txt","NI_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/NI_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto/intermediates/TWAS/T2D/T2D.txt","Flu_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/Flu_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/T2D/T2D.txt","NI_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/NI_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/T2D/T2D.txt","Flu_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/Flu_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/T2D/T2D.txt","NI_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/NI_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/T2D/T2D.txt","Flu_standard":"/beagle3/haky/users/saideep/projects/aracena_modeling/SPrediXcan/standard_predictDB_aracena/Flu_T2D_filtered.txt","NI_standard":"/beagle3/haky/users/saideep/projects/aracena_modeling/SPrediXcan/standard_predictDB_aracena/NI_T2D_filtered.txt", "GTeX_WB": "/beagle3/haky/users/saideep/projects/aracena_modeling/SPrediXcan/GTex_WB/T2D_SPrediXcan_results_GTex_WB_trimmed.txt" },"AE": {"Flu_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/Flu_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto/intermediates/TWAS/Allergy_Eczema/Allergy_Eczema.txt","NI_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/NI_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto/intermediates/TWAS/Allergy_Eczema/Allergy_Eczema.txt","Flu_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/Flu_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/Allergy_Eczema/Allergy_Eczema.txt","NI_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/NI_ws4_ns_3_7_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/Allergy_Eczema/Allergy_Eczema.txt","Flu_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/Flu_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/Allergy_Eczema/Allergy_Eczema.txt","NI_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1":"/beagle3/haky/users/saideep/projects/Con_EnPACT/models/NI_ws4_ns_4_4_lnormTRUE_beforeTRUE_scale1/intermediates/TWAS/Allergy_Eczema/Allergy_Eczema.txt","Flu_standard":"/beagle3/haky/users/saideep/projects/aracena_modeling/SPrediXcan/standard_predictDB_aracena/Flu_AE_Farreira_filtered.txt","NI_standard":"/beagle3/haky/users/saideep/projects/aracena_modeling/SPrediXcan/standard_predictDB_aracena/NI_AE_Farreira_filtered.txt","GTeX_WB":"/beagle3/haky/users/saideep/projects/aracena_modeling/SPrediXcan/GTex_WB/AE_SPrediXcan_results_GTex_WB_trimmed.txt" }}# "GTeX_WB", # gwas_traits = ["T2D", "AE"]# expression_models = ["Flu_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto", # "NI_ws4_ns_3_7_lnormFALSE_beforeFALSE_scale1_sourcekallisto", "GTeX_WB"]# path_to_results = "/beagle3/haky/users/saideep/projects/Con_EnPACT/SPrediXcan_results"# for trait in gwas_traits:# for model in expression_models:# input_file = os.path.join(path_to_results, trait, model, f"{trait}_SPrediXcan_results_{model}.csv")# if not os.path.exists(input_file):# print(f"File {input_file} does not exist")# sys.exit(1)
Plotting functions
Code
import pandas as pdimport scipy.stats as statsdef to_latent(rgb_non_latent, alpha=0.5):return [x/255.0for x in rgb_non_latent] + [alpha]def load_results(input_files, tag=False, only_tag=False, model_contains_string=[""], model_excludes_string=[""]): output_dict = {}for gwas_type in input_files.keys(): output_dict[gwas_type] = {}for model_type in input_files[gwas_type].keys():# Check if model name contains all substrings does_mcs_all =Trueiftype(model_contains_string) ==str: model_contains_string = [model_contains_string]for mcs in model_contains_string:if mcs notin model_type: does_mcs_all =Falsebreakifnot does_mcs_all:print("Does not contain all substrings")continue# Check if model name contains any exclusion substrings does_mes_any =Falseiftype(model_excludes_string) ==str: model_excludes_string = [model_excludes_string]for mes in model_excludes_string:if mes =="":continueif mes in model_type: does_mes_any =Truebreakif does_mes_any:print("Contains exclusion substring")continue# Check if tagged datasets should be loadedif only_tag:passelse: cur_df = pd.read_csv(input_files[gwas_type][model_type], sep=",") output_dict[gwas_type][model_type] = cur_dfif tag:iftype(tag) ==str: cur_df = pd.read_csv(input_files[gwas_type][model_type].replace(".txt", "_"+tag+".txt"), sep=",") cur_df_no_na = cur_df.dropna(subset=["pvalue"]) output_dict[gwas_type][model_type+"_"+tag] = cur_df_no_naelse:for t in tag: cur_df = pd.read_csv(input_files[gwas_type][model_type].replace(".txt", "_"+t+".txt"), sep=",") cur_df_no_na = cur_df.dropna(subset=["pvalue"]) output_dict[gwas_type][model_type+"_"+t] = cur_df_no_nareturn output_dict# Create function for creating subsetted tables def create_gene_subsets_SPrediXcan_results(gene_table, tag, SPrediXcan_results_files, gene_table_col="ensembl"): loaded_results = load_results(SPrediXcan_results_files)for gwas in SPrediXcan_results_files.keys():for model in SPrediXcan_results_files[gwas].keys(): cur_df = loaded_results[gwas][model] cur_df_subset = cur_df[cur_df['gene'].isin(gene_table[gene_table_col])]# print("subsetting")# print(cur_df_subset.shape)# print(sum(cur_df_subset['gene'].isin(gene_table[gene_table_col])))ifnot SPrediXcan_results_files[gwas][model].endswith(".txt"):print("Error: File does not end with .txt")return cur_df_subset.to_csv(SPrediXcan_results_files[gwas][model].replace(".txt", "_"+tag+".txt"), sep=",", index=False)
Code
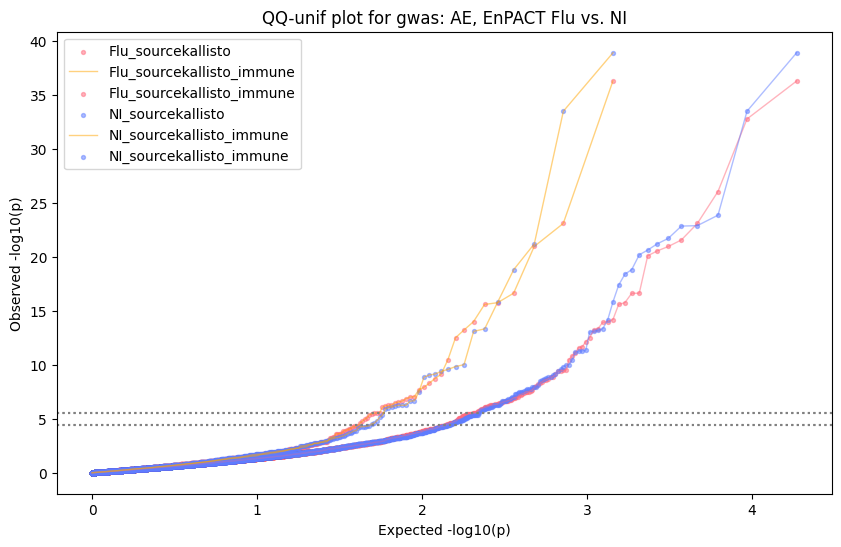
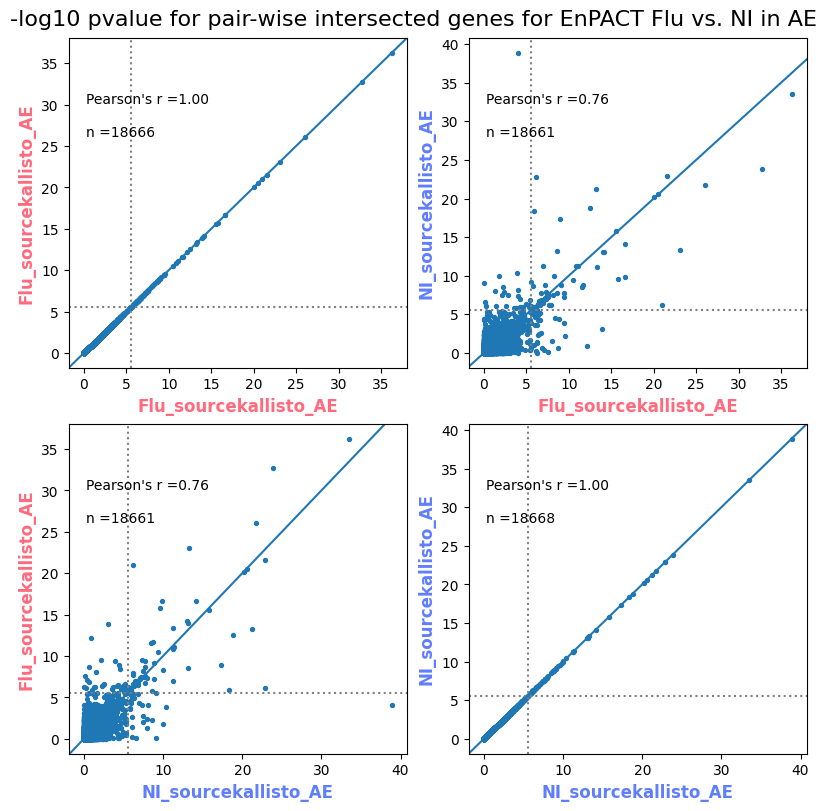
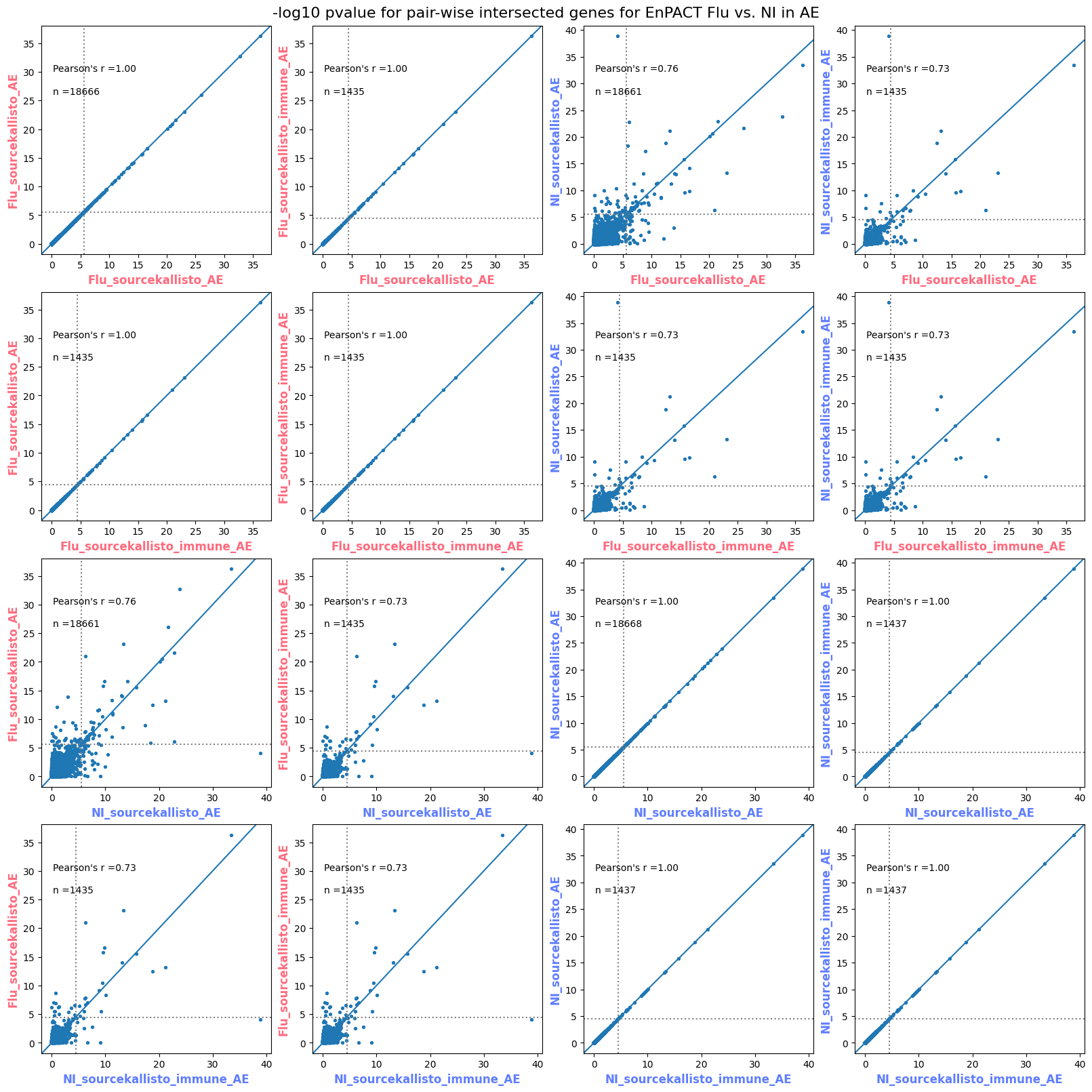
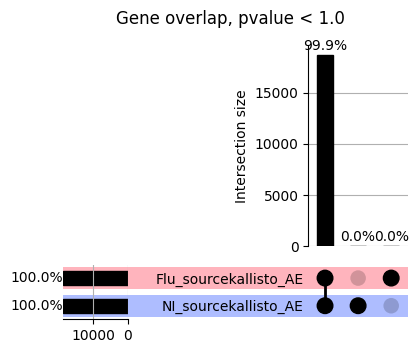
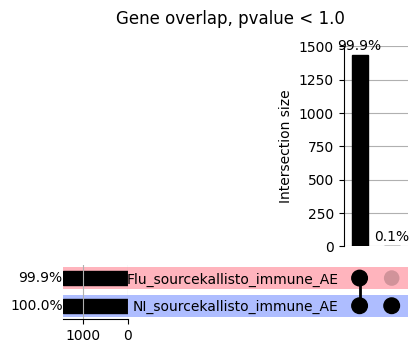
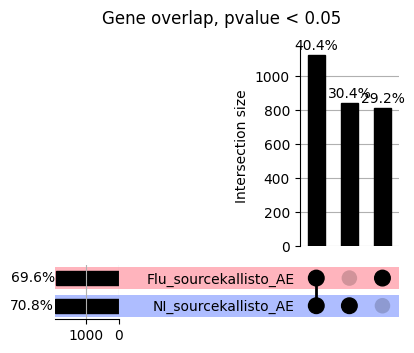
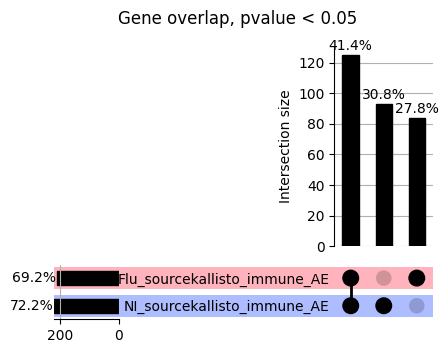
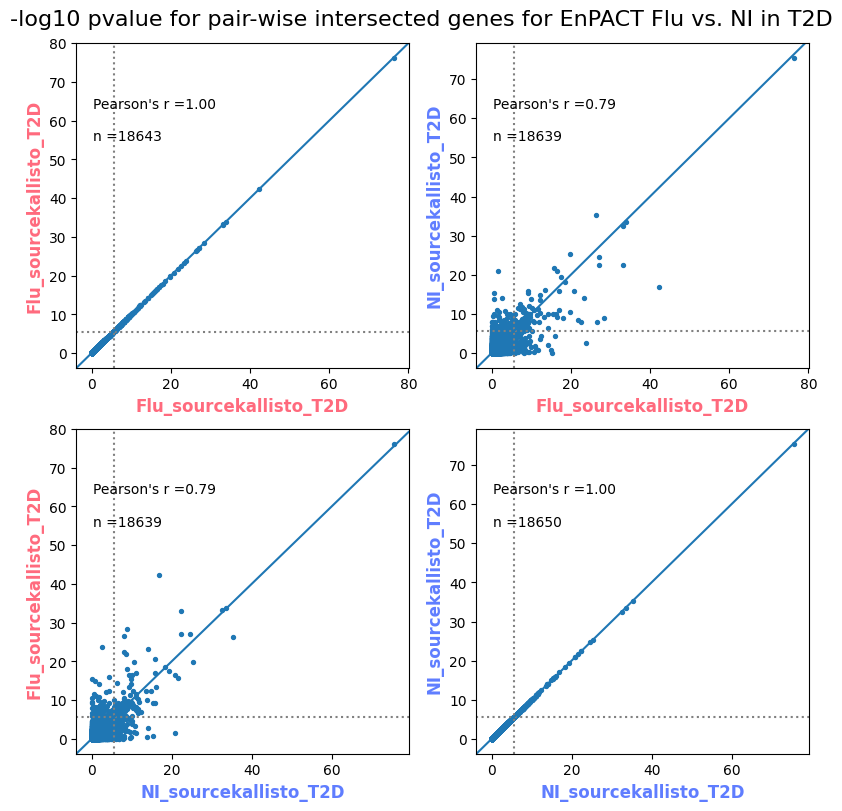
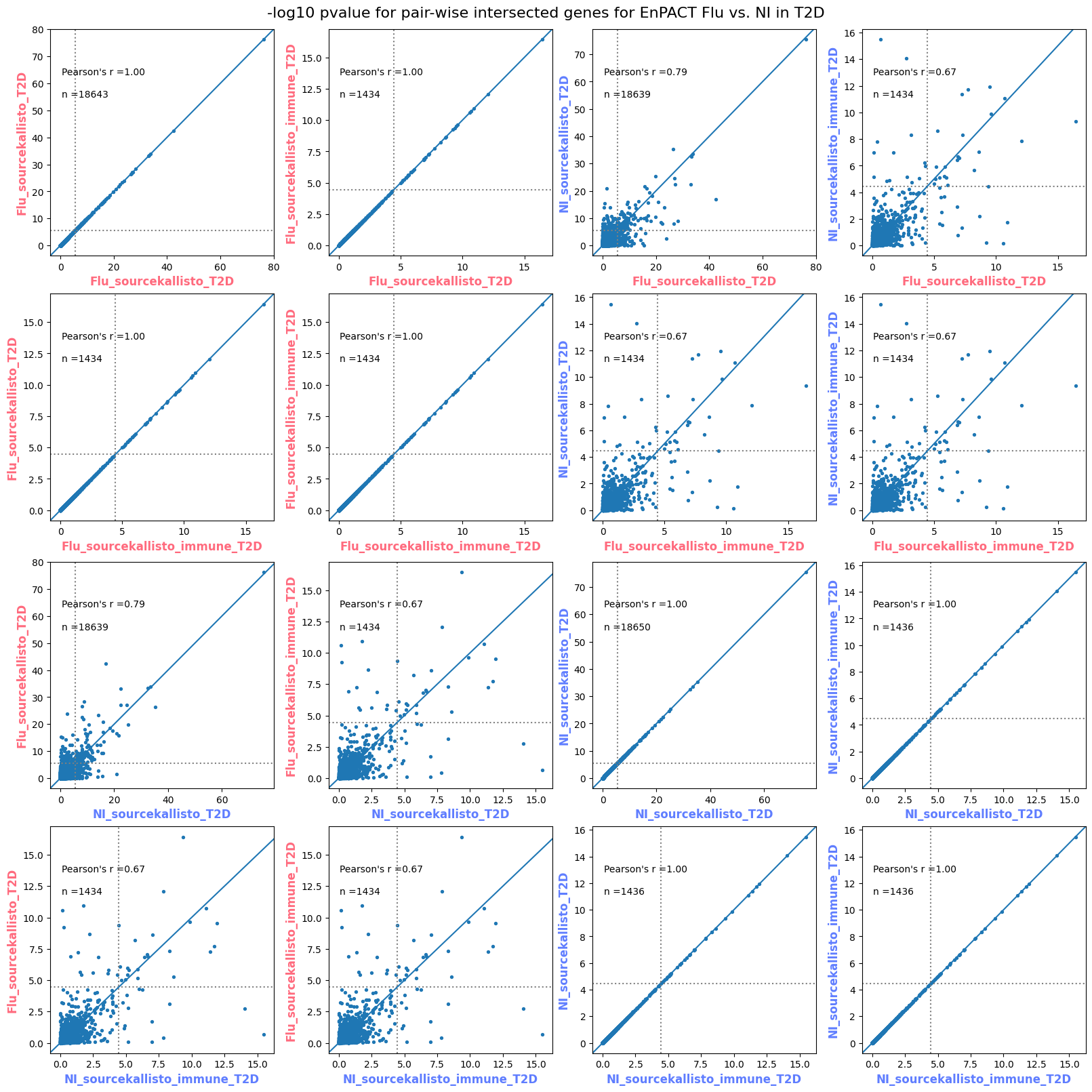
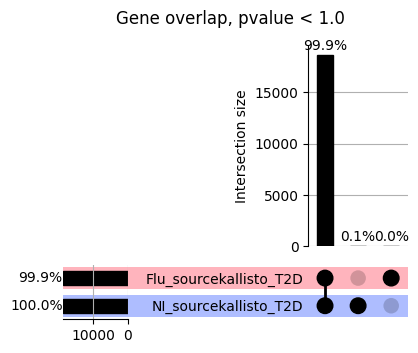
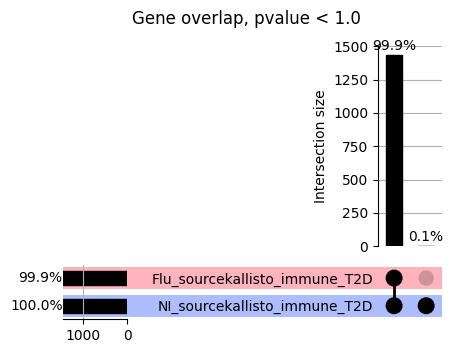
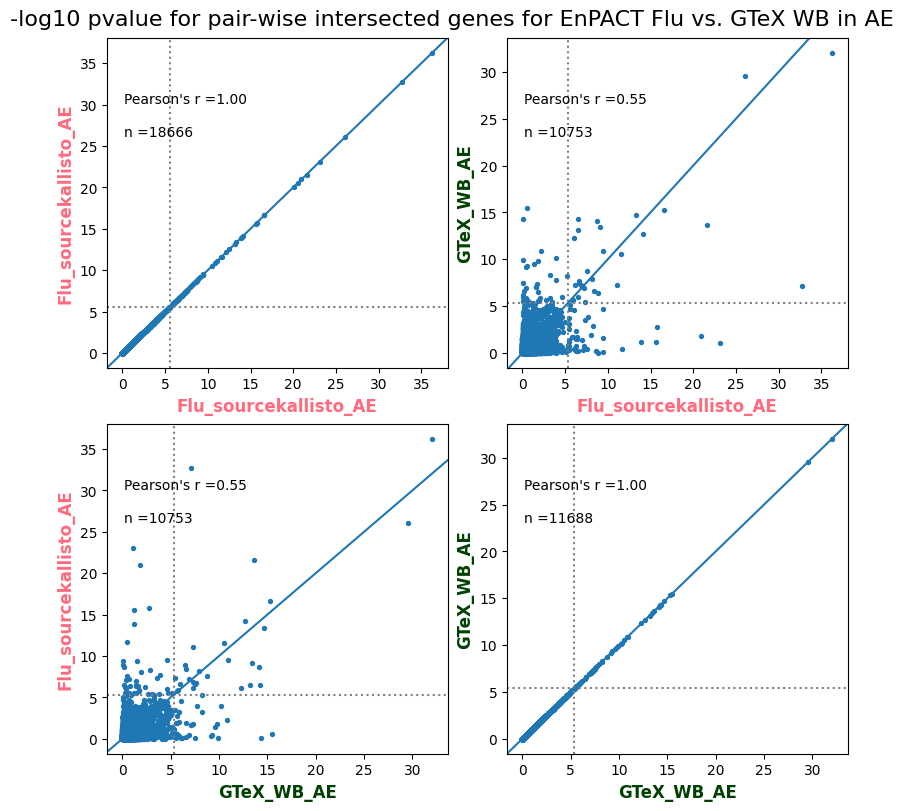
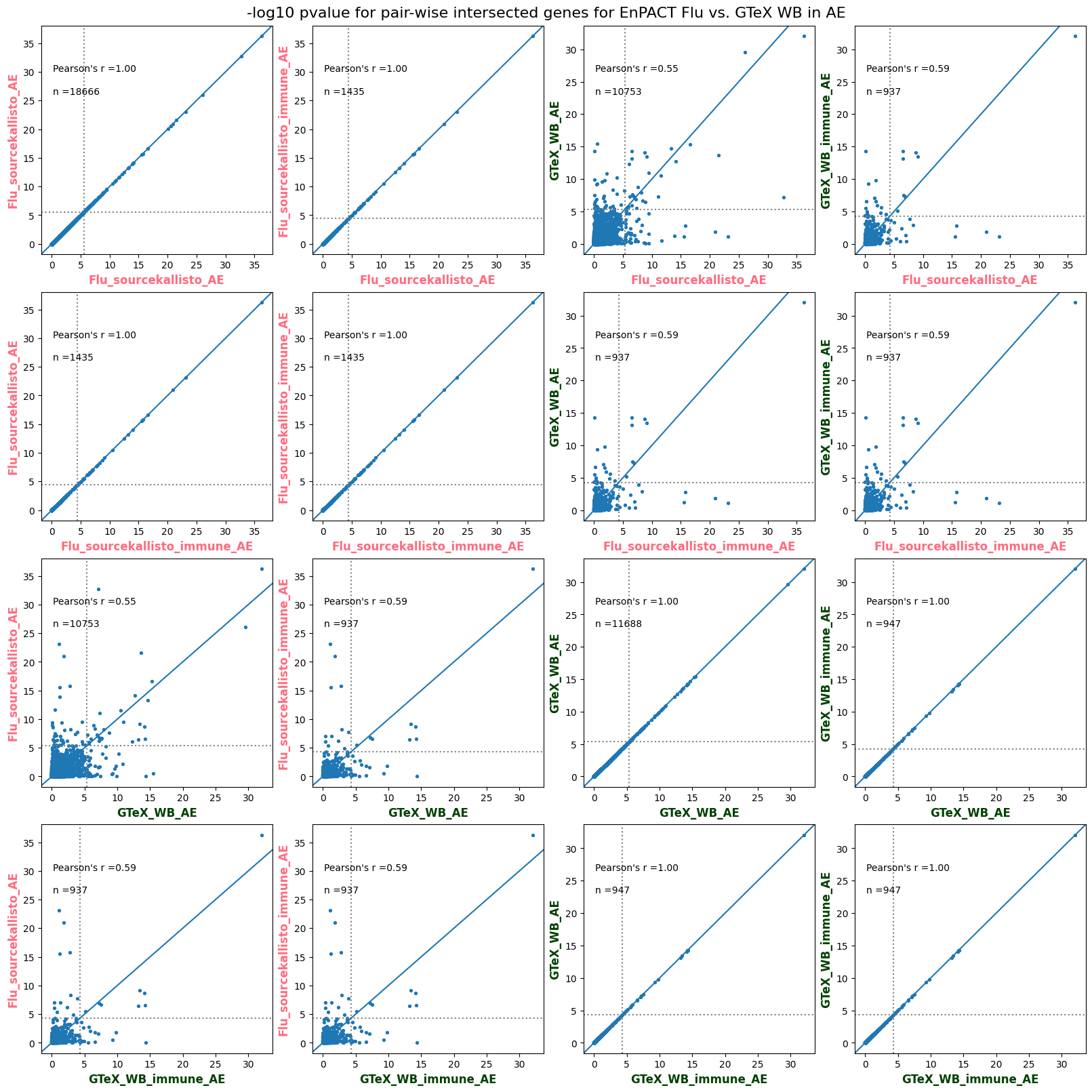
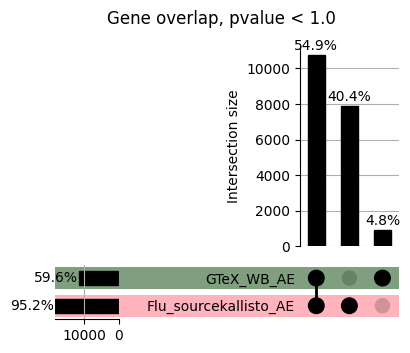
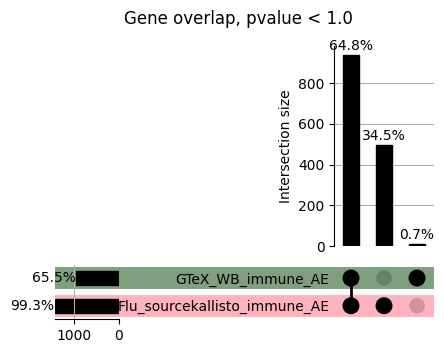
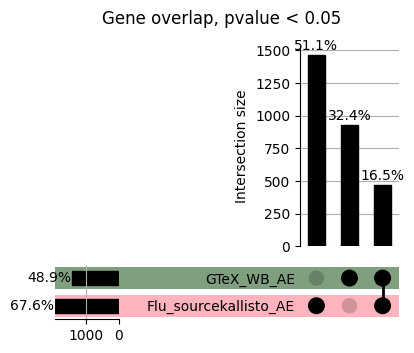
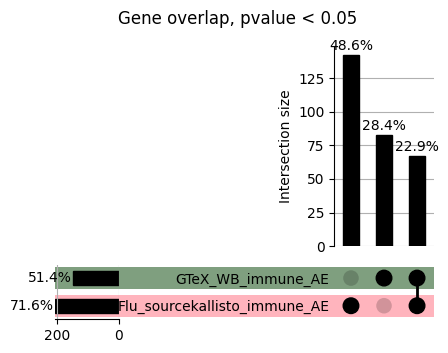
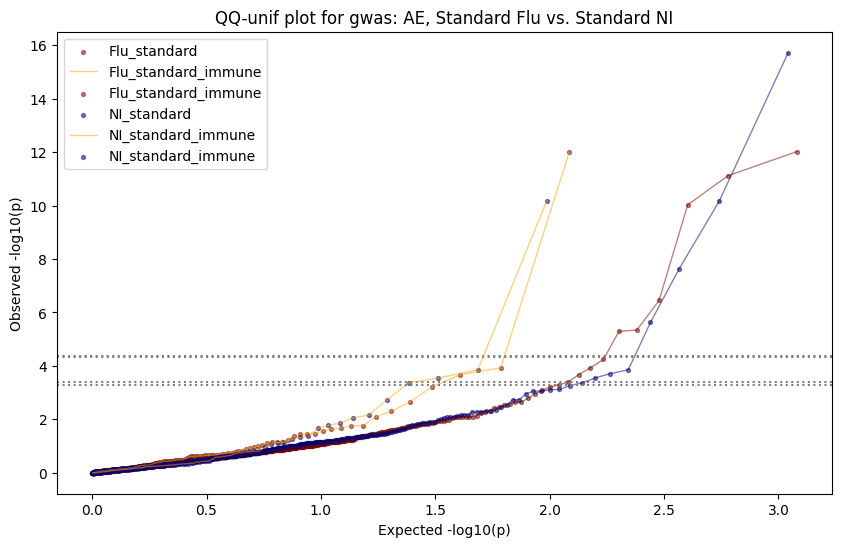
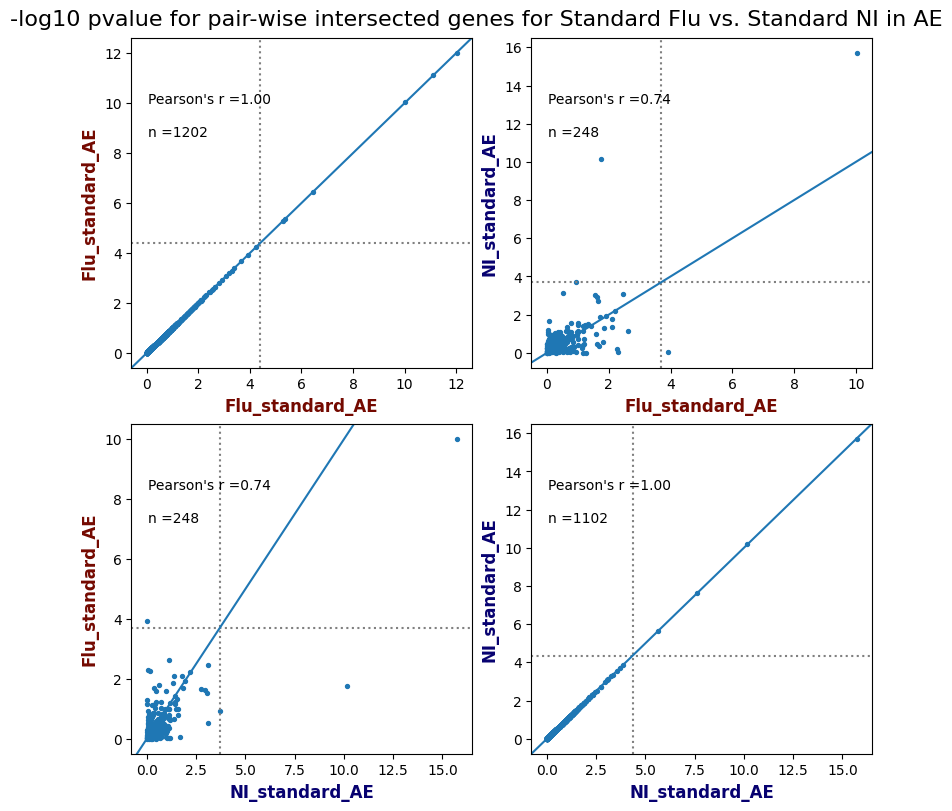
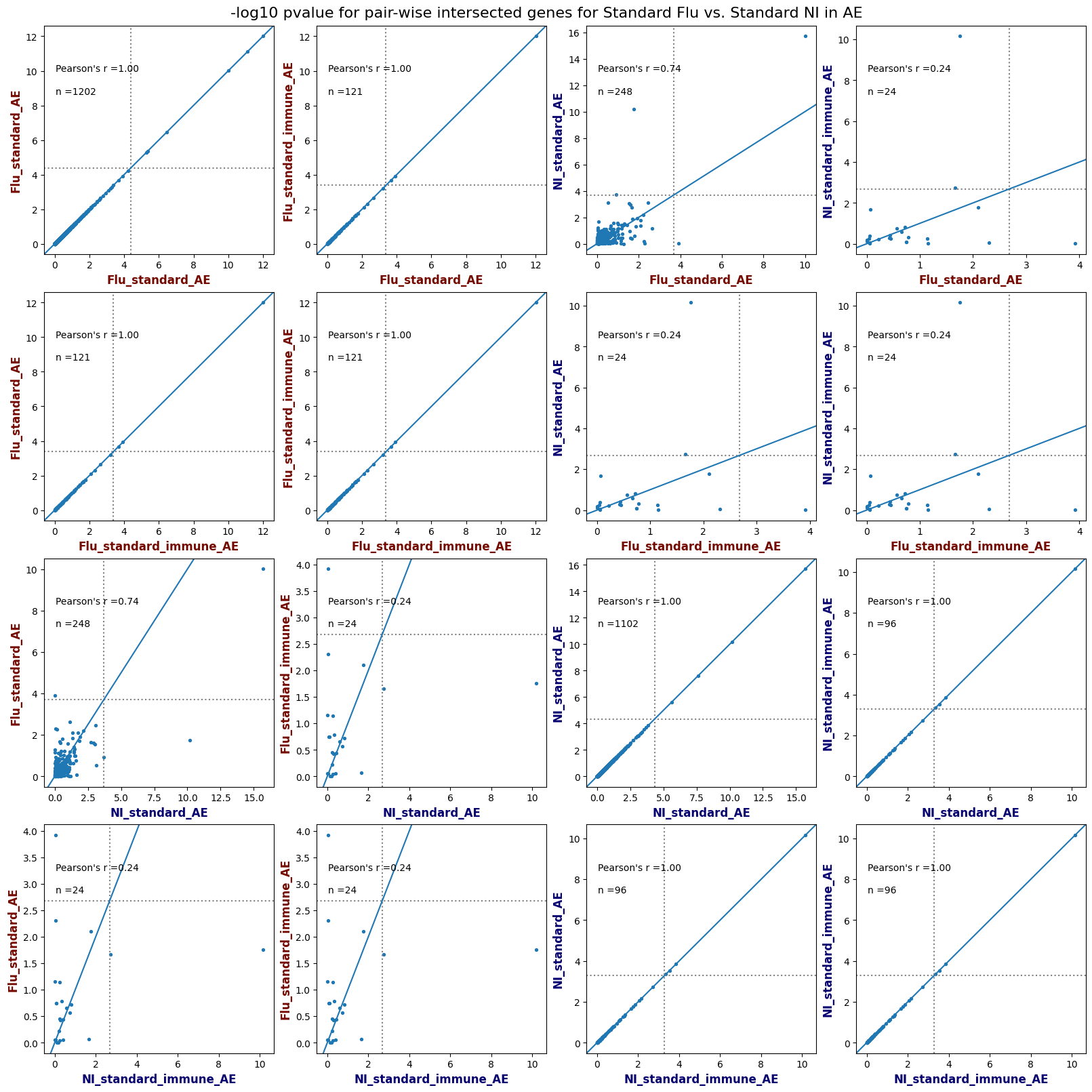
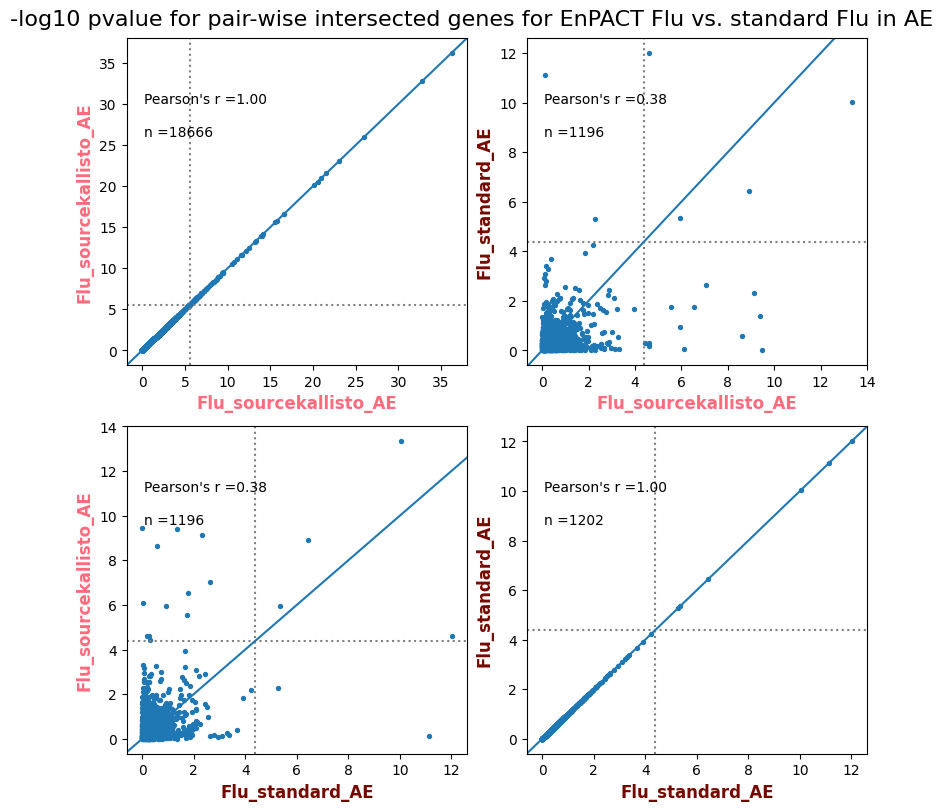
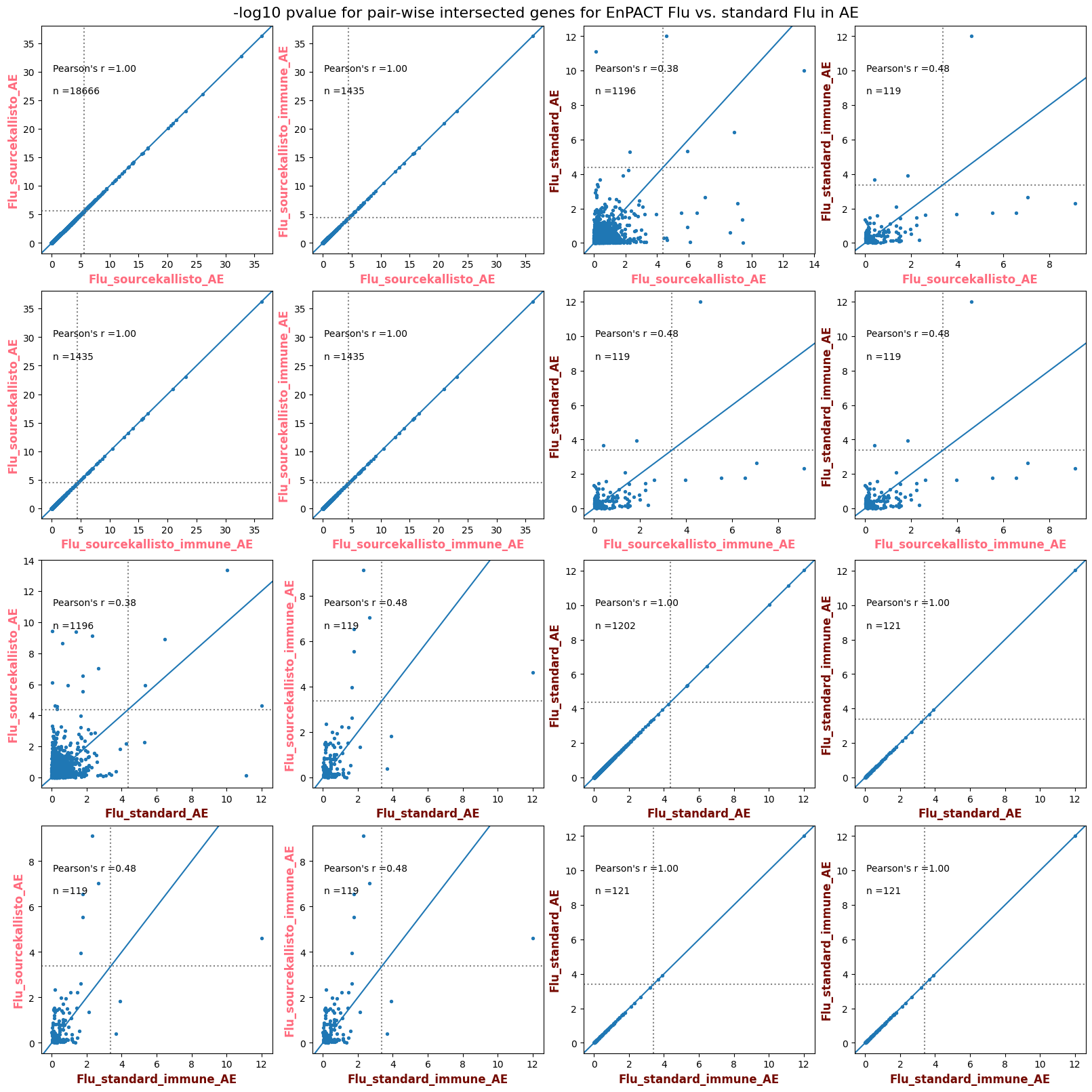
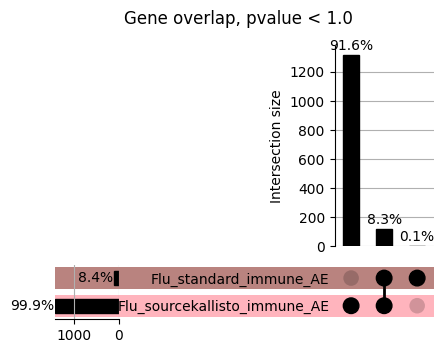
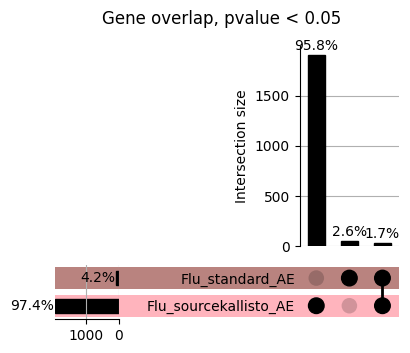
import matplotlib.pyplot as pltimport numpy as npfrom scipy.stats import betaimport upsetplotdef qqunif_maxp(p, label, BF=False, CI=False, mlog10_p_thres=30, maxp=1, color=False, lw=1.0): nn =len(p) xx =-np.log10((np.arange(1, nn +1) * maxp) / (nn +1))ifnot color: plt.scatter(xx, -np.sort(np.log10(p)), s =8, label=label) plt.plot(xx, -np.sort(np.log10(p)), lw=lw)else:if"secondary"in color: plt.plot(xx, -np.sort(np.log10(p)), lw=lw, label=label, color=to_latent(color["secondary"]))else: plt.plot(xx, -np.sort(np.log10(p)), lw=lw, color=to_latent(color["primary"])) plt.scatter(xx, -np.sort(np.log10(p)), s =8, label=label, color=to_latent(color["primary"]))if BF: plt.axhline(y=-np.log10(0.05/nn), linestyle=':', color='grey')if CI: c95 = np.zeros(nn) c05 = np.zeros(nn)for i inrange(1, nn +1): c95[i -1] = beta.ppf(0.95, i, nn - i +1) * maxp c05[i -1] = beta.ppf(0.05, i, nn - i +1) * maxp plt.plot(xx, -np.log10(c95)) plt.plot(xx, -np.log10(c05))def qq_plot_unif(S_results, GWAS, save_dir, colors=False, BF=False, CI=False, title_tag ="", plot_filename="default"): plt.figure(figsize=(10, 6))# plt.axhline(-np.log10(0.05), color='red', linestyle='-', label='FDR = 0.05')# plt.axhline(-np.log10(0.10), color='orange', linestyle='--', label='FDR = 0.10')# plt.axhline(-np.log10(0.25), color='yellow', linestyle='-.', label='FDR = 0.25')for S_result in S_results.keys():ifnot colors: qqunif_maxp(S_results[S_result].pvalue, label=f'{S_result}', BF=BF, CI=CI)else: qqunif_maxp(S_results[S_result].pvalue, label=f'{S_result}', BF=BF, CI=CI, color=colors[S_result]) plt.xlabel("Expected -log10(p)") plt.ylabel("Observed -log10(p)") plt.title(f"QQ-unif plot for gwas: {GWAS}, "+title_tag) plt.legend()if plot_filename =="default": plt.savefig(os.path.join(save_dir,f'{GWAS}_qqplot.png'))else: plt.savefig(os.path.join(save_dir,plot_filename)) plt.show()def hist_plot(S_results, GWAS, model, save_dir): plt.figure(figsize=(10, 6)) plt.hist(S_results.pvalue, bins=100) plt.xlabel("Expected -log10(p)") plt.ylabel("Observed -log10(p)") plt.title(f"QQ-unif plot for {GWAS},{model}") plt.legend() plt.savefig(os.path.join(save_dir,f'{GWAS}_{model}_histogram.png')) plt.show()def scatter_plots(S_results):# Assumes equal rows in all dataframesfor S_result in S_results.keys(): dict_for_plot = {}for model in S_results[S_result].keys(): sorted_cur_df = S_results[S_result][model].sort_values(by=["gene"]) dict_for_plot[model] =list(sorted_cur_df["pvalue"]) df_for_plot = pd.DataFrame(dict_for_plot) pd.plotting.scatter_matrix(df_for_plot, figsize=(len(S_results[S_result].keys())*5,len(S_results[S_result].keys())*5), diagonal='kde')def create_subset_df(list_of_dfs, subset_col="gene", join="inner"): subset_df = list_of_dfs[0].copy()for df in list_of_dfs[1:]: subset_df = subset_df.merge(df, how=join,on=subset_col)return subset_dfdef print_dims(results_dict):for gwas_type in results_dict.keys():for model_type in results_dict[gwas_type].keys():print(f"{gwas_type}{model_type} : {results_dict[gwas_type][model_type].shape}")def upset_plot(loaded_results, pval_thresh=0.05, substrings=[""], exclude_substrings=[""], percent=True, colors=False): contents = {}for gwas_type in loaded_results.keys():for model_type in loaded_results[gwas_type].keys():# Check if substrings are present in keywords all_substrings_present =Truefor substring in substrings:if substring notin (model_type+"_"+gwas_type): all_substrings_present =Falseifnot all_substrings_present:continue# Check if substrings for exclusion are present in keywords exclude_substring =Falsefor exclude_sub in exclude_substrings:if exclude_sub =="":continueif exclude_sub in (model_type+"_"+gwas_type): exclude_substring =Trueif exclude_substring:continue# Filter by pvalue cur_df = loaded_results[gwas_type][model_type] cur_df_filt = cur_df.loc[cur_df["pvalue"]<pval_thresh] cur_gene_list =list(cur_df_filt["gene"]) contents[model_type+"_"+gwas_type] = cur_gene_list contents_for_plot = upsetplot.from_contents(contents)try:if percent:ifnot colors: u = upsetplot.UpSet(contents_for_plot, orientation='horizontal', sort_by='cardinality', show_percentages=True, )else: u = upsetplot.UpSet(contents_for_plot, orientation='horizontal', sort_by='cardinality', show_percentages=True )for twas_type in contents.keys(): twas_type_wo_gwas ="_".join(twas_type.split("_")[0:-1]) u.style_categories(twas_type, shading_facecolor=to_latent(colors[twas_type_wo_gwas]["primary"]))else:ifnot colors: u = upsetplot.UpSet(contents_for_plot, orientation='horizontal', sort_by='cardinality', show_counts=True, )else: u = upsetplot.UpSet(contents_for_plot, orientation='horizontal', sort_by='cardinality', show_counts=True, )for twas_type in contents.keys(): twas_type_wo_gwas ="_".join(twas_type.split("_")[0:-1]) u.style_categories(twas_type, shading_facecolor=to_latent(colors[twas_type_wo_gwas]["primary"])) u.plot() plt.suptitle("Gene overlap, pvalue < "+str(pval_thresh)) plt.show()exceptExceptionas e:print(e)def scatter_plot(first_df, second_df, ax, x_lab="x", y_lab="y", sort_by="gene", bf=True, colors=False): common_genes =list(set(first_df["gene"]).intersection(set(second_df["gene"]))) first_df_subset = first_df[first_df["gene"].isin(common_genes)].sort_values(by=[sort_by]) second_df_subset = second_df[second_df["gene"].isin(common_genes)].sort_values(by=[sort_by]) nan_filt = np.logical_not(np.logical_or(np.isnan(-np.log10(list(first_df_subset["pvalue"]))), np.isnan(-np.log10(list(second_df_subset["pvalue"]))))) r,p = stats.pearsonr(x=-np.log10(first_df_subset[nan_filt]["pvalue"]),y=-np.log10(second_df_subset[nan_filt]["pvalue"])) n =len(first_df_subset[nan_filt]["pvalue"]) ax.scatter(-np.log10(first_df_subset[nan_filt]["pvalue"]), -np.log10(second_df_subset[nan_filt]["pvalue"]), s=8)if bf: ax.axhline(y=-np.log10(0.05/n), linestyle=':', color='grey') ax.axvline(x=-np.log10(0.05/n), linestyle=':', color='grey') ax.text(.05, .8, "Pearson's r ={:.2f}".format(r), transform=ax.transAxes) ax.text(.05, .7, f"n ={n}", transform=ax.transAxes) ax.set_xlabel(x_lab, fontsize=12, weight ='bold') ax.set_ylabel(y_lab, fontsize=12, weight ='bold')ifnot colors:passelse: ax.xaxis.label.set_color(to_latent(colors["_".join(x_lab.split("_")[0:-1])]["primary"], alpha=1.0)) ax.yaxis.label.set_color(to_latent(colors["_".join(y_lab.split("_")[0:-1])]["primary"], alpha=1.0)) ax.axline((0, 0), slope=1)def scatter_plot_all(results_dict, plot_title="-log10 pvalue for pair-wise intersected genes", colors=False): results_count =0for gwas_type in results_dict.keys():for model_type in results_dict[gwas_type].keys(): results_count +=1 fig = plt.figure(figsize=(4*results_count, 4*results_count), layout="constrained") fig.suptitle(plot_title, fontsize=16) spec = fig.add_gridspec(ncols=results_count, nrows=results_count) ind1 =0 ind2 =0for gwas_type1 in results_dict.keys():for model_type1 in results_dict[gwas_type].keys(): ind2 =0for gwas_type2 in results_dict.keys():for model_type2 in results_dict[gwas_type].keys(): ax = fig.add_subplot(spec[ind1, ind2]) scatter_plot(results_dict[gwas_type1][model_type1], results_dict[gwas_type2][model_type2], ax, x_lab=model_type1+"_"+gwas_type1, y_lab=model_type2+"_"+gwas_type2, colors=colors) ind2 +=1 ind2 =0 ind1 +=1
# for gwas in loaded_results.keys():# for model in loaded_results[gwas].keys():# pass# hist_plot(loaded_results[gwas][model], gwas, model, "/beagle3/haky/users/saideep/github_repos/Daily-Blog-Sai/posts/2023-12-06-analyze_SPrediXcan_results/plots")
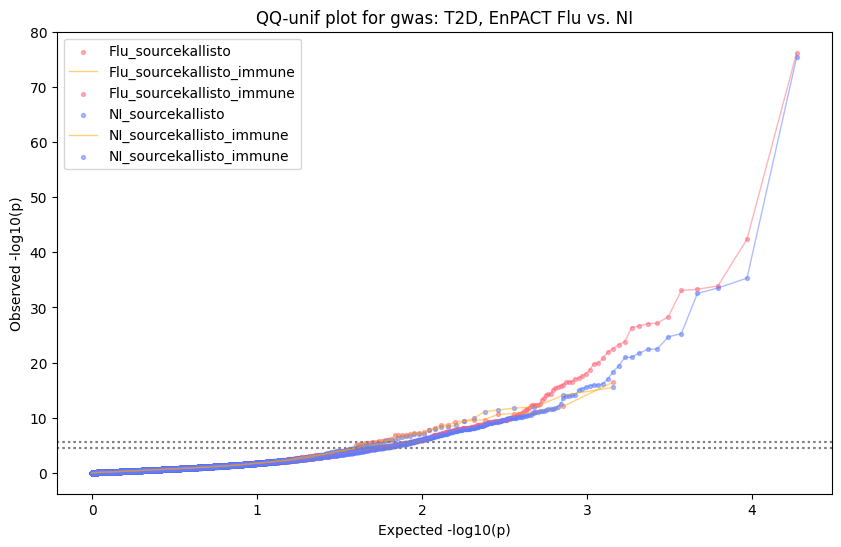
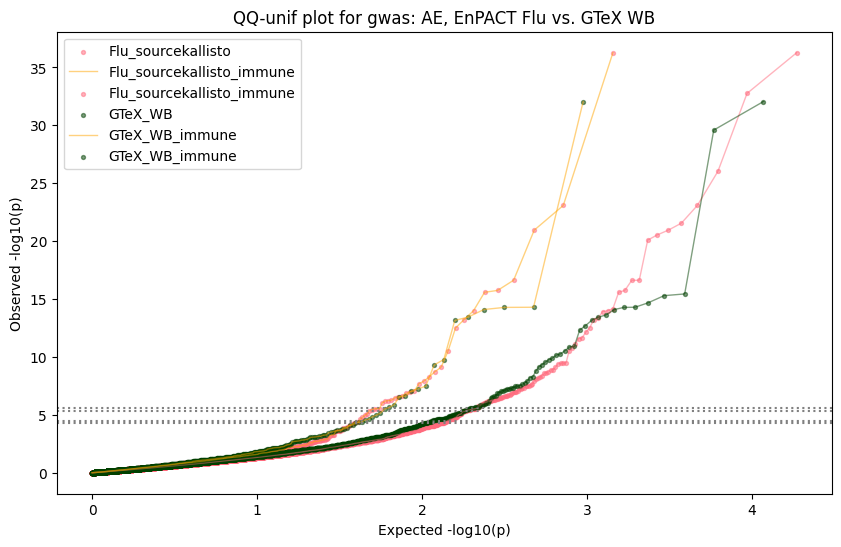
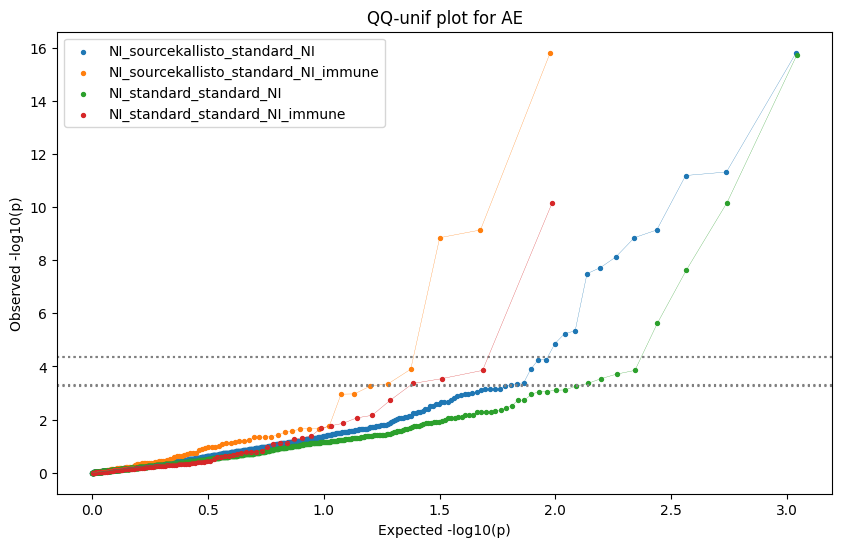
Generate qqplots
Investigating immune-related genes
The gene list here is from the IRIS database of immune-related genes: https://pubmed.ncbi.nlm.nih.gov/15780753/ and downloaded from: https://www.innatedb.com/moleculeSearch.do
Code
# Load in immune gene listpath_to_immune_genes ="/beagle3/haky/users/saideep/github_repos/Daily-Blog-Sai/posts/2023-12-06-analyze_SPrediXcan_results/InnateDB_genes.csv"immune_genes = pd.read_csv(path_to_immune_genes, sep=",")immune_genes.head()# Also look at common Flu-NI genes# flu_genes = loaded_results['T2D']['Enpact_Flu']# ni_genes = loaded_results['T2D']['Enpact_NI']# list_of_dfs = [flu_genes, ni_genes]# flu_ni_subset = create_subset_df([flu_genes, ni_genes], subset_col="gene")# print(flu_genes.shape, ni_genes.shape)# flu_ni_subset.shape
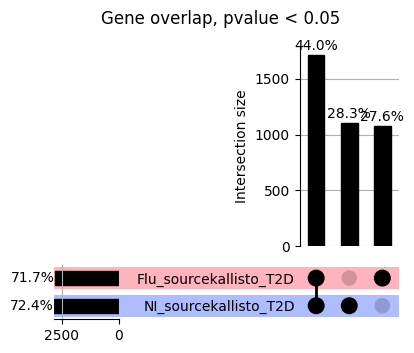
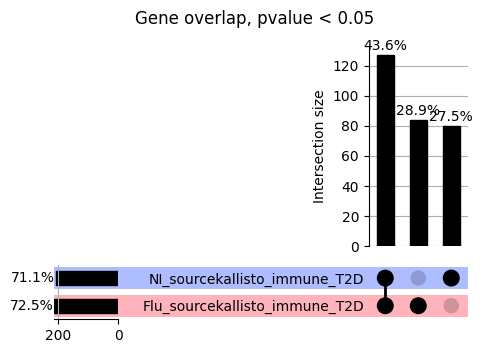
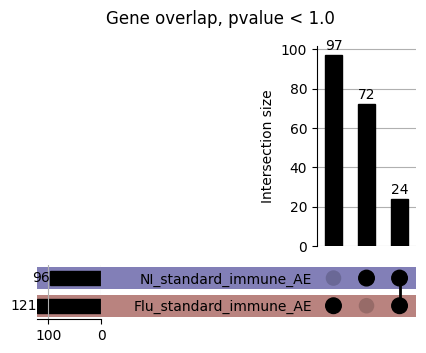
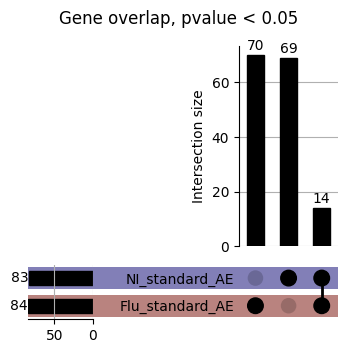
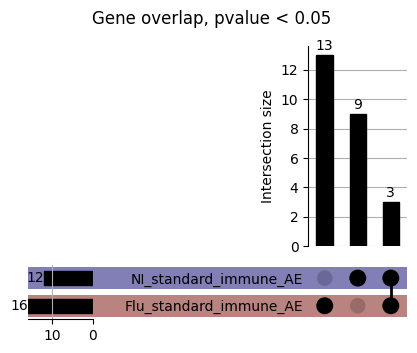
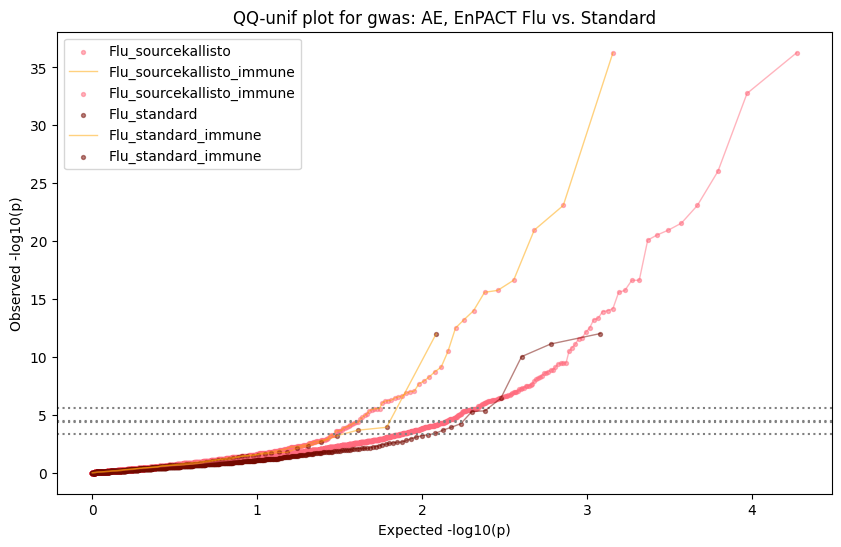
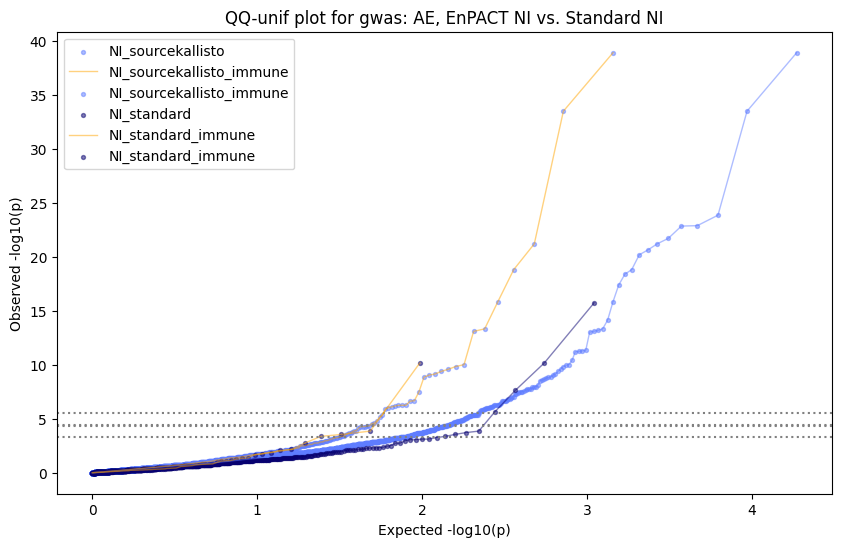
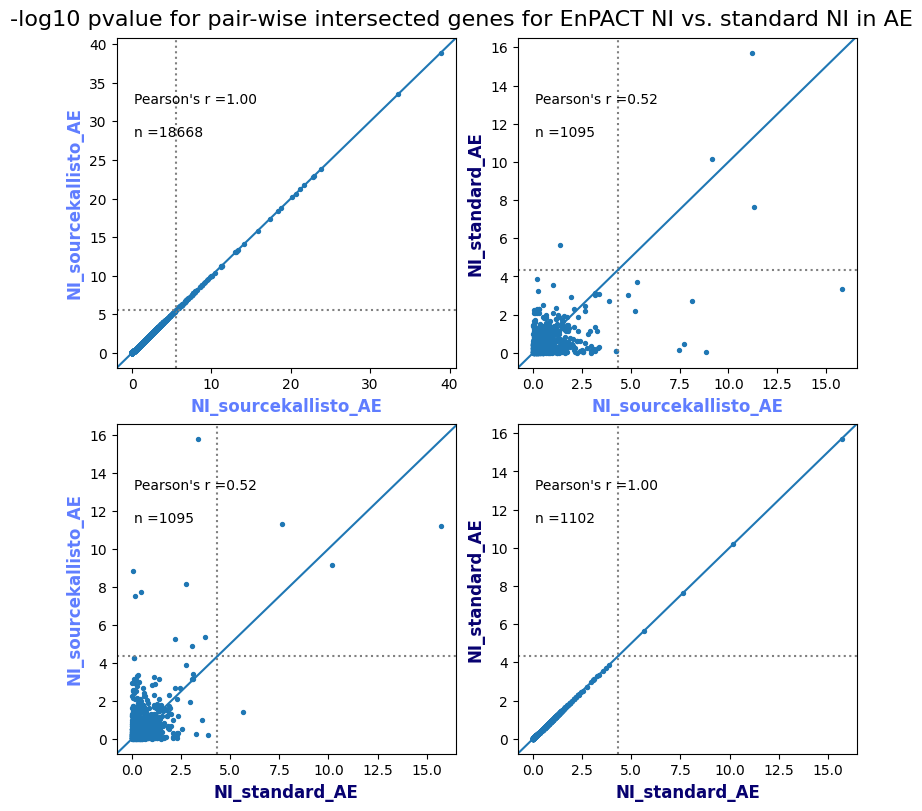
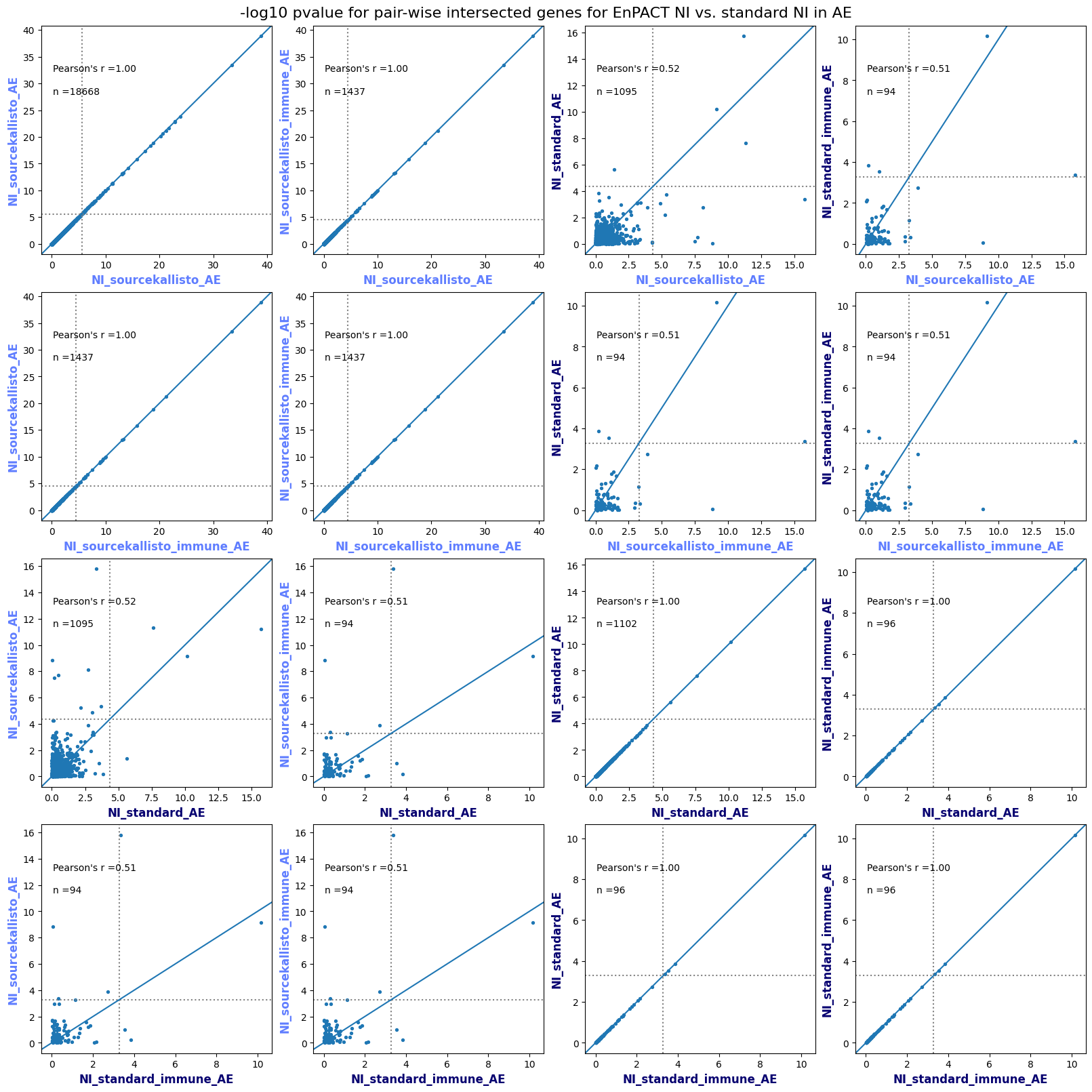
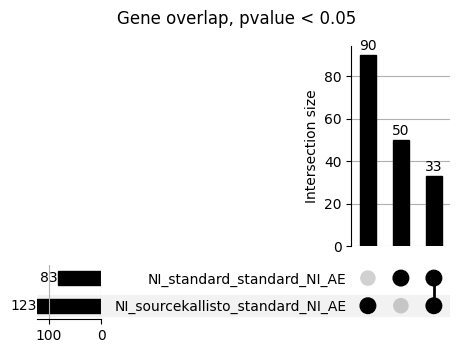
How does EnPACT compare with Standard PrediXcan in NI condition?
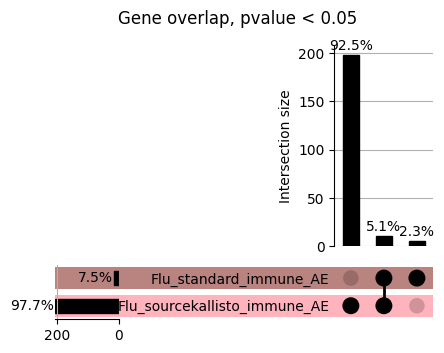
Code
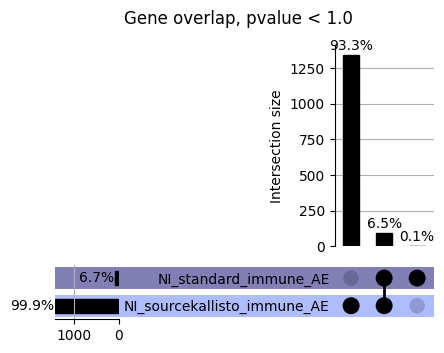
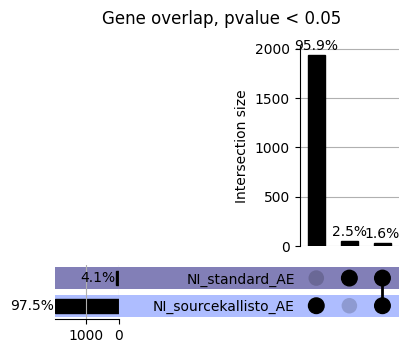
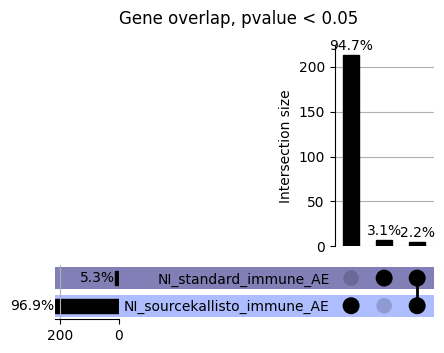
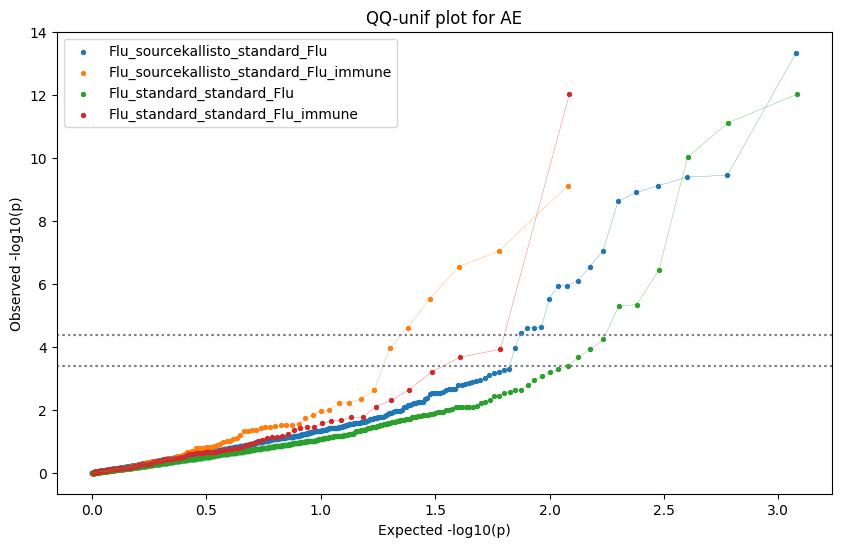
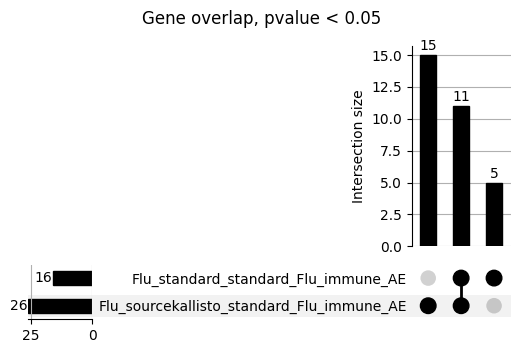
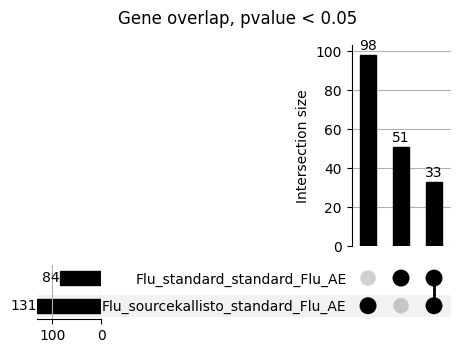
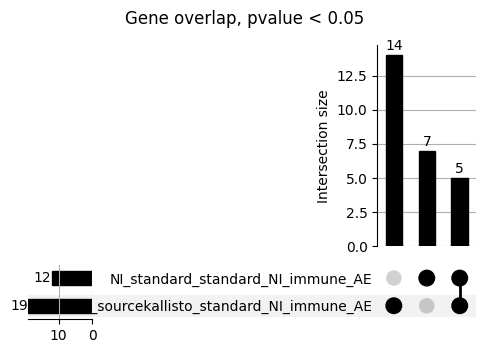
loaded_results_compare_standard_NI = load_results(input_files_compare_standard, tag=["immune"], model_excludes_string="Flu")qq_plot_unif(loaded_results_compare_standard_NI[gwas], gwas, "/beagle3/haky/users/saideep/github_repos/Daily-Blog-Sai/posts/2023-12-06-analyze_SPrediXcan_results/plots", plot_filename=f"compare_standard_qqplot.png", BF=True, title_tag="EnPACT NI vs. Standard NI", colors=color_pal_compare_standard)scatter_plot_all(load_results(input_files_compare_standard, model_excludes_string="Flu"), colors=color_pal_compare_standard, plot_title=f"-log10 pvalue for pair-wise intersected genes for EnPACT NI vs. standard NI in {gwas}")scatter_plot_all(loaded_results_compare_standard_NI, colors=color_pal_compare_standard, plot_title=f"-log10 pvalue for pair-wise intersected genes for EnPACT NI vs. standard NI in {gwas}")upset_plot(loaded_results_compare_standard_NI, pval_thresh=1.0, substrings=["immune", "NI"], percent=False, colors=color_pal_compare_standard)upset_plot(loaded_results_compare_standard_NI, pval_thresh=0.05, substrings=["NI"], exclude_substrings=["immune"], percent=False, colors=color_pal_compare_standard)upset_plot(loaded_results_compare_standard_NI, pval_thresh=0.05, substrings=["immune", "NI"], percent=False, colors=color_pal_compare_standard)